Article topic: Neuronal Storage Diseases
Author: Nour Nedal Al-Bzour
Editors: Bashar Abualsiba’, Sadeen Eid
Reviewer: Ethar Hazaimeh
Keywords: Neuronal storage diseases, lysosomal storage diseases, Tay-Sachs, rare diseases, CNS, NSD, LSD, Gaucher, Fabry
Introduction
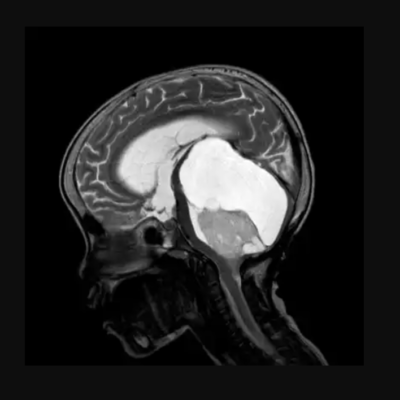
Neuronal Storage Diseases (aka Lysosomal Storage Diseases (LSDs)) are chronic rare inherited metabolic diseases. LSDs are caused by a deficiency of the lysosomal enzymes that are needed to catabolize substances in the cells of the central nervous system (CNS)¹. This enzymatic deficiency causes accumulation of the substrates in the lysosome and thus initiates a cascade of cellular events including inflammatory and apoptotic responses. The accumulation and storage of these substrates may begin in early embryonic development and causes inborn metabolic errors.² Although LSDs are considered rare diseases, the prevalence of LSDs is 1 in 2000.³
History
LSDs are a set of diseases in which there is a storage of substrates in the lysosomes. Some of them develop prenatally, directly after birth, or in childhood or adulthood. The first disease discovered in this set was Gaucher in 1882, followed by Fabry in 1898. The diseases are classified according to the type of substrate being accumulated. For example, Fabry’s disorder results from the accumulation of glycosphingolipid globotriaosylceramide in the lysosome and can progressively develops and lead to organ failure and resulting in premature death.4
Epidemiology
LSDs are considered rare when studying them solely, yet when they are combined with each other, they are much more common. Many factors increase the incidence rate of developing LSDs, these include ethnicity and geography. For example, there is a higher incidence rate of Gaucher’s disease in Jews (Eastern European Jews), it can be as high as 1 in 800, likewise, in Tay-Sachs disease, the chance of developing the disease is 1 in 3900.5
Types and Symptoms
LSDs contain more than 70 inherited disorders. Although the signs and symptoms vary from group to group, they share the main cause behind the disease which is an enzymatic deficiency.
Table 1 summarizes some of these disorders and their characteristics
| Disease | Clinical features |
| Galactosialidosis | Cerebellar ataxia, myoclonus, visual failure; onset in late childhood or adolescence
|
| GM1-gangliosidosis | Coarse facial features, corneal clouding, hepatomegaly, skeletal dysplasia, learning disability, myoclonus
|
| Infantile-free sialic acid storage disease (ISSD) | Coarse facies, fair complexion, hepatosplenomegaly, severe psychomotor retardation, nephrotic syndrome
|
| Tay-Sachs disease | Exaggerated acoustic motor reflex,
Megalocephaly
|
| Sandhoff disease | Exaggerated acoustic motor reflex, splenomegaly
|
| Niemann-Pick disease type A | Hepatosplenomegaly, spasticity
|
| Neuronal ceroid lipofuscinosis (NCL) | Cerebellar ataxia, myoclonus, visual failure, onset by late childhood or adolescence |
Table 1: Lysosomal Storage Diseases types and characteristics
Diagnosis
For a definitive diagnosis of LSDs, advanced laboratory biochemical and genetics are required. The diagnosis of LSDs has been confirmed by assaying specific deficient enzymes or accumulated substrates using serum, white blood cells, urine, amniotic fluid, and cultured skin fibroblasts.6 Some secondary changes may occur in cells with a lysosomal deficiency which may lead to modifications of proteins and other cellular components that can be a useful biomarker for the detection and diagnosis of LSDs.7
The tests used to diagnose LSDs can be classified into six categories, the most common of which are;
- Urinary oligosaccharides
This test is performed by high-performance thin-layer chromatography. However, this technique has some limitations. Metabolite quantification is not possible, and the identification of metabolites is assumed based on migration rates. Because of these limitations, this technique can be used as an initial diagnosis for LSDs, but further investigations will be needed to confirm the diagnosis. - Urinary glycosaminoglycans (GAGs)
Mucopolysaccharidoses which is a type of LSDs can be suspected by the measurements of urinary GAGs. First, the GAGs are centrifuged and isolated, then quantified by colorimetric assay. The pitfall of this technique is the progressive decrease of urinary GAGs that occurs with age. Thus, the normal-age decline in urinary GAGs should be taken into consideration when diagnosing LSDs.9 - Global Tests
It has been found that LAMP-1, LAMP-2, saposins, and GM2-ganglioside are elevated in almost all LSDs. 10
Treatments
Back in history, there was no treatment used for LSDs. The aim back then was to alleviate the pain and reduce the symptoms without working on the underlying pathogenesis. Other treatments used before had bad outcomes, such as Gaucher disease, bone marrow transplantation proved to be successful treatment although with a mortality rate of 10%. Same as in Fabry’s disease, renal translation turned out to be successful but cannot correct the multi-organ manifestations of the disease. Enzymatic therapy was then introduced, first used for the treatment of Gaucher disease, then Fabry, mucopolysaccharidosis type I (MPS I), MPS VI, and Pompe disease, and is under development for MPS II (Hunter syndrome).11
The currently available treatments for LSDs have some limitations. Neurodegenerative disease in LSDs is challenging to treat, and the efficient treatments available do not meet the patients’ needs. Thus, a combination of early diagnosis and therapeutic approaches may be required. A prudent approach is to prevent lysosomal storage, increase lysosomal degradation (autophagy), increase lysosomal exocytosis, and enhance lysosomal function.
LSDs treatment, as well as targeted therapy methods, are mentioned in table 2 below :
| Treatment Target | Potential Therapy | Lysosomal Storage Diseases in which the Therapy was Trailed |
| Aberrant activation inflammation | Pentosan polysulfate | MPS I, II In vitro in MPS I, IIIA, VI |
| Intraperitoneal high-dose aspirin | MPS IIIB | |
| Miglustat | Niemann–Pick disease, type C1 | |
| Increase cellular energy | Resveratrol | Neuronal ceroid lipofuscinosis |
| Autophagic pathway | – | In vitro in Pompe disease |
| Arimocromol | Niemann–Pick disease, type C1 | |
| – | Krabbe disease | |
| Reduction in mitochondrial ROS generation | Coenzyme Q10 | Gaucher disease + Fabry disease |
| GSH ethyl-ester | Niemann–Pick disease, type C1 | |
| N-acetylcysteine | Krabbe disease Niemann–Pick disease, type C1 |
|
| Lysosomal calcium signaling | Curcumin | Niemann–Pick disease, type C1 |
| Diltiazem | Gaucher disease | |
| mTOR signaling/transcription factor TFEB | Rapamycin | Multiple Sulfatase Deficiency |
| Apoptotic pathway | Z-DEVD-FMK | Niemann–Pick disease, type C1 |
| Z-VAD-FMK | GM1 gangliosides | |
| Modulation of sitrulins | Resveratrol | Infantile NCL |
| Inhibition of GSK-3-signalling | L803-mts | Krabbe disease |
Table 2: LSDs treatment and targeted therapy12
Future Directions
Therapeutic treatments under investigation include hematopoietic stem cell transplantation (HSCT) with autologous cells that have been modified genetically to express supra-physiological enzyme levels. Moreover, recombinant enzyme trials administered intravenously and intrathecally have been undertaken or are ongoing.13 In addition, gene therapy with oligodendroglia, neural progenitor, and embryonic and microencapsulated recombinant cells is being investigated as well.14
Conclusions
LSDs are chronic and can affect the quality of life and lead to shortened lifespans, especially with neuropathic involvement. Pathophysiological factors such as neurodegeneration, neuroinflammation, impaired autophagy, and microgliosis are major causes of progressive neurogenerative decline in patients with neurological LSD. Our knowledge and understanding of the structure and function of the lysosomes are still developing, and there is still needed for more information and studies to find more efficient therapeutic strategies for treating LSDs.
References...