Article topic: Parkinson’s Disease
Author: Kafa Al-Zoubi
Editors: Yazan S. Al-Zamer, Ethar Hazaimeh
Keywords: Parkinson’s disease (PD), PARK7 (Parkinsonism Associated Deglycase), LRRK2 (leucine-rich repeat kinase 2), PINK1 (PTENinduced putative kinase 1), SNA( alpha-synuclein)
Overview
Parkinson’s disease(PD) is a chronic progressive neurodegenerative movement disease with early prominent death of dopaminergic neurons in the substantia nigra pars compacta (SNpc) (1), The second most common disease of neurodegeneration after Alzheimer’s disease is Parkinson’s disease(2). The disease was first identified in 1817 by a British physician named James Parkinson who described it in “An Essay on the Shaking Palsy”(3).
As a consequence of the dopamine deficit in the basal ganglia lead to movement disorder(4). PD is characterized by four cardinal signs, resting tremor, rigidity, bradykinesia, and postural instability, and they’re all motor symptoms(5). PD also involves nonmotor symptoms such as depression, sleep disturbances, and orthostatic hypotension(6). It’s associated with severe disability and a negative impact on quality of life.
The prevalence of PD is 0.3% in the general population, 1.0% in people older than 60 years, and 3.0% in people older than 80 years, and incidence rates of PD are estimated to range between 8 and 18 per 100,000 person-years(7). The vast majority of Parkinson’s disease cases are sporadic, with just 10% of patients having a disease-causing genetic mutation. There are significant sex-related differences in epidemiological and clinical characteristics of the disease(8). Men are twice as likely as women to develop Parkinson’s disease(9), but women have a higher mortality rate and a rapid disease progression(10).In addition, as compared to men, women show different symptoms as well as differences in response to pharmacological treatments and deep brain stimulation procedures, as well as in personal assessments of the quality of life(11).
Etiology
PD is a multifactorial disorder that involves both genetic and environmental factors. Around 80% of people with Parkinson’s disease are considered idiopathic, while the remaining 20% are considered to be genetic(12). The most important risk factor for Parkinson’s disease is age, with the median age of onset being 60 years old (13).
Environmental factors:
- Cigarette smoking
Several studies demonstrate that there is an inverse association between smoking and PD (14). They also observed an inverse relationship between the number of pack-years, years of smoking, and the risk of PD. Compared to nonsmokers, heavy or long-term smokers have a significantly lower chance of developing Parkinson’s disease (15). The reasons underlying this association are not well understood. But, activation of nicotinic acetylcholine receptors on dopaminergic neurons by nicotine or selective agonists is neuroprotective in experimental models of PD(16).
- Caffeine
It has been reported that coffee drinkers have a 25% lower chance of developing PD (17). Caffeine is an adenosine receptor antagonist, which is believed to be protective in PD (18). This association is weak in females because estrogen competitively inhibits caffeine metabolism and in post-menopausal women that depend on hormone replacement therapy(19).
- Pesticides, herbicides, and heavy metals
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is metabolized into the neurotoxin, MPP+ (1-methyl-4-phenylpyridinium) which is a mitochondrial complex-I inhibitor that selectively destroys dopaminergic cells in the substantia nigra(20). The discovery of MPTP as a cause of nigral degeneration led to the hypothesis that PD could be caused by an environmental toxin such as Paraquat (a herbicide) and rotenone (a pesticide) are also selective complex-I inhibitors and induce dopaminergic depletion in animal models of PD (21).
Genetics factor
- Autosomal recessive
Parkin (PARK2) was the first gene detected that causes an early-onset autosomal recessive type of PD before 40 years old(22). It is located on chromosome 6q25.2–q27 and encodes an E3 ubiquitin ligase involved in the cellular ubiquitination-protein degradation pathway(23). As a consequence of this gene mutation leading to loss of function of the protein, impacting many different pathways, including the ubiquitin-proteasome system degradation pathway, oxidative/dopaminergic stress, and mitochondrial function. In addition, it does not associate with the Lewy body(24).
PINK1 (PARK6) encodes 581‑amino acid protein which includes a mitochondrial-targeting motif and a highly conserved protein kinase domain with homology to the serine/threonine kinases of the calcium/calmodulin family (25). PINK1 is steady on mitochondria with lower membrane potential and it recruits Parkin from the cytosol Parkin becomes enzymatically active and initiates the autophagic clearance of mitochondria through lysosomes(26).
- Autosomal dominant
SNCA (PARK1–4), the gene encoding for a-synuclein, was the first gene related to autosomal-dominant Parkinson’s disease and A53T was the first pathogenic SNCA mutation detected(27) that result in a higher inclination to a-synuclein to misfolded and aggregate. Other pathogenic mutations affect the quantity of a-synuclein (either through duplications or triplications) and alter its post-transcriptional modifications or its interaction with other cellular organelles and transport systems(7).
LRRK2 (PARK8) mutations are the most common known cause of late-onset autosomal-dominant and encode the 2527-amino acid (28). It contains The LRRK2 protein contains putative GTPase, protein kinase, WD40 repeat, and leucine-rich repeat (LRR) domains of unidentified functions(29). LRRK2 mutations are associated with dopaminergic neuronal loss and gliosis in the substantia nigra(30).
Pathogenesis
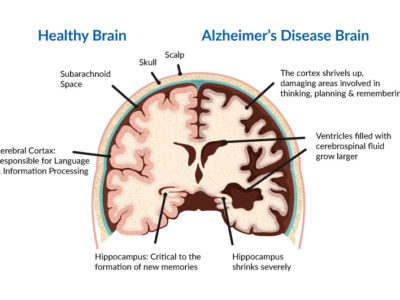
The main pathological features of PD are the absence of dopaminergic neurons followed by depigmentation of the SNpc and the presence of LB.LBs are intraneuronal, round, eosinophilic inclusions with a hyaline core and a pale peripheral halo that contains more than 90 proteins mainly a-synuclein and ubiquitin(31)(32).
Oxidative stress
Oxidative stress has obtained the most attention in PD due to the capacity of the oxidative metabolism of dopamine to yield hydrogen peroxide (H2O2) and other reactive oxygen species(33). Oxidant stress and consequent cell death could develop in the SNc underneath circumstances in which there are elevated dopamine turnover, leading to excess peroxide formation, a deficiency in glutathione (GSH), thereby diminishing the brain’s capability to clean H2O, or an increase in reactive iron, that may promote OH formation (34).
Studies have confirmed that iron levels are elevated inside the substantia nigra of PD patients and it accumulates mostly within neuromelanin granules of dopaminergic neurons (33). How iron accumulates inside the SNc in PD is not recognized. Elevated lactoferrin receptors were detected on nigral neurons in PD patients and can account for preferential accumulation of iron inside those cells(35). It is not clear if iron accumulation in PD is primary or secondary. In addition, elevated SNc iron has been observed following MPTP treatment or 6-hydroxydopamine (6-OHDA) lesions. These results indicate that iron may accumulate secondary to cell degeneration caused by many factors and still cause cell death(36).
In Parkinson’s disease, a deficiency in one or more of the antioxidant defenses could lead to neurodegeneration(37). Reduction in GSH can inhibit H2O2 clearance and promote OH formation mainly in the presence of elevated iron. The cause of GSH deficiency in PD is unclear(33).
There is an elevated in the level of γ -glutamyltranspeptidase (γ -GTT), the enzyme responsible for the translocation of glutathione precursors and metabolism of the oxidized form of glutathione and this is elevation may reflect an attempt by surviving cells to recruit glutathione precursors into the cell to replenish reduced levels of GSH or a compensatory mechanism to remove potentially toxic GSSG produced as a consequence of oxidant stress(38).
There is evidence of oxidative damage in the brains of PD patients. The lipid peroxidation products malondialdehyde (MDA) and lipid hydroperoxide have been found higher in the SNc of PD patients, but not in the cerebellum(39). In surviving dopaminergic neurons, increased staining for 4-hydroxynonenal, a product of lipid peroxidation that can modify proteins and induce cell toxicity(40). Increased amounts of protein carbonyls and 8-hydroxy-2-deoxyguanosine, which indicate oxidative damage to proteins and DNA, have also been discovered in the SNc and other brain regions of PD patients(41).
Mitochondrial Dysfunction
Studies demonstrated that in the SNc of PD patients there is a reduction in complex I function of the mitochondrial respiratory chain for unclear reasons (42). A mitochondrial complex I defect could lead to cell degeneration in PD through reduced ATP synthesis and a bioenergetic defect. A mitochondrial complex I defect could also lead to cell damage through free radicals generated directly on-site or as a result of a compensatory increase in respiration at complex Ⅱ. A complex Ⅰ defect can also play a role in the onset of apoptosis. Since complex Ⅰ is the primary site of proton pumping, a complex Ⅰ defect in PD could cause neuronal weakness and apoptosis. A reduction in the mitochondrial membrane potential caused by defective proton pumping can lead to the opening of a mitochondrial permeability transition pore and the release of small mitochondrial proteins that signal for the decrease in mitochondrial membrane potential caused by defective proton pumping may result in the opening of a mitochondrial permeability transfer pore and the release of small mitochondrial proteins that signal the beginning of apoptosis(33).
Neuroinflammatory markers
Neuroinflammation is the CNS’s inflammatory reaction to toxic stimuli, which disrupts neuronal homeostasis and leads to a variety of neurological disorders (43). Neuroinflammation in PD arises alongside the loss of dopaminergic neurons and is related to altered microglia activity in of CNS and other parts. Recent studies have found the presence of activated microglia either inside substantia nigra (SN) and putamen of patients with a PD prognosis(44). After activating microglia, tumor necrosis factor (TNF-) releases a variety of free radicals and activates many genes and proteins, including iNOS and proinflammatory cytokines(IL-1β, IL-6, and α)(45).
Overexpression of the central immune system may compromise the production of neurotrophic factors and cytotoxic factors from the microglia cell. IL-1β family of cytokines that is a pro-inflammatory molecule also activates toll-like receptors (TLRs), which may disrupt the clearance of microglia. studies show that disrupting IRAK-4 (interleukin-1 receptor-associated kinase 4), an essential downstream kinase of TLRs and receptors for IL-1β, moves microglia cells from a pro-inflammatory phenotype toward an anti-inflammatory phenotype(46).