Article Topic: Amyotrophic Lateral Sclerosis (ALS)
Author: Boshra Alajarma
Keywords: Neurons, Frontotemporal Dementia, Free Radicals, Motor Muscle
Scientific Editor: Marah Bassam Akhdar
Linguistic Editor: Noor AlOsta
Introduction
This disease has many names, such as; Lou Gehrig’s disease, Motor Neuron Disease (MND) and Charcot disease. Amyotrophic Lateral Sclerosis (ALS) is a set of rare neurological diseases that mainly affects the neurons responsible for controlling voluntary muscle movement such as the muscles responsible for chewing food, speaking and walking.
This disease is progressive. Motor neuron loss continues until the ability to eat, speak, move, and breathe is lost. Most people with ALS die within 3 to 5 years from when the first symptom appears, because of respiratory failure. However, only about 10 percent of people with ALS survive for 10 or more years[1]. ALS results from the death of lower motor neurons in the spinal cord and brain stem as well as upper motor neurons (Betz cells) in the motor cortex[7].
Although the primary symptoms of ALS are associated with motor dysfunction as muscle weakness, spasticity and dysphagia, up to 50% of patients develop cognitive and/or behavioral impairment, and 13% of patients present with concomitant behavioral variant frontotemporal dementia (FTD) during the course of the disease[2].
Epidemiology
ALS is the most common motor neuron disease in adults and the third most common neurodegenerative disease[3]. The number of new cases a year in Europe is 2.6 people per 100,000 with the number of affected people being 7-9 per 100,000. In the United States in 2015,there were 5.2 people per 100,000 diagnosed with ALS. In Addition, it was higher in white people, males, and those over 60 years of age[5]. Also, there are differences in the genetics of ALS between different ethnic groups. The most common ALS gene in Europe is C9orf72, then SOD1, TARDBP, and FUS. However, the most common ALS gene in Asia is SOD1, followed by FUS, C9orf72, and TARDBP[6].
Pathogenesis
Most cases of ALS are sporadic and about 10% of the cases are familial, mostly with autosomal dominant inheritance. Familial diseases begin earlier in life than sporadic diseases. However, once the symptoms appear, the clinical course is similar in both forms[7]. The basic feature of ALS is the death of both upper motor neurons located in the motor cortex of the brain and lower motor neurons located in the brainstem and spinal cord. In people who suffer from ALS with frontotemporal dementia, neurons throughout the frontal and temporal lobes of the brain die. The pathological hallmark of ALS is the presence of inclusion bodies, which are abnormal aggregations of bunina bodies (proteins in the cytoplasm of motor neurons) [14].
Scientists have identified 25 genes to be associated with Amyotrophic Lateral Sclerosis (ALS). The pathogenic mutation in the C9orf72 gene is a hexanucleotide repeat expansion [GGGGCC]. The C9orf72 gene is the most commonly associated gene with ALS, accounting for 40% of familial ALS and 7% of sporadic ALS cases[8].
Superoxide Dismutase Gene (SOD1) on chromosome 21 was the first identified genetic cause of ALS and accounts for about 20% of the familial forms. These mutations are thought to generate abnormal misfolded forms of the SOD1 protein which may trigger the unfolded protein response and cause apoptotic death of neurons[7]. Superoxide Dismutase (SOD1) enzyme is a powerful antioxidant that protects the body from damage caused by superoxide, which is a toxic free radical generated in the mitochondria. During normal metabolism, cells generate free radicals which are highly reactive molecules, those free radicals can cause damage to the DNA and proteins within cells. To date, over 110 different mutations in SOD1 have been linked with the disorder, some of which as H46R have a very long clinical course, while others such as A4V are exceptionally aggressive. When the defenses against oxidative stress fail, apoptosis is upregulated. Moreover, up to 180 different mutations in SOD1 gene are known to cause familial ALS[9].
TARDBP, which codes for TAR DNA-binding protein (TDP-43), was found to be associated with 1–5% of familial ALS and less than 1% of sporadic ALS[10].
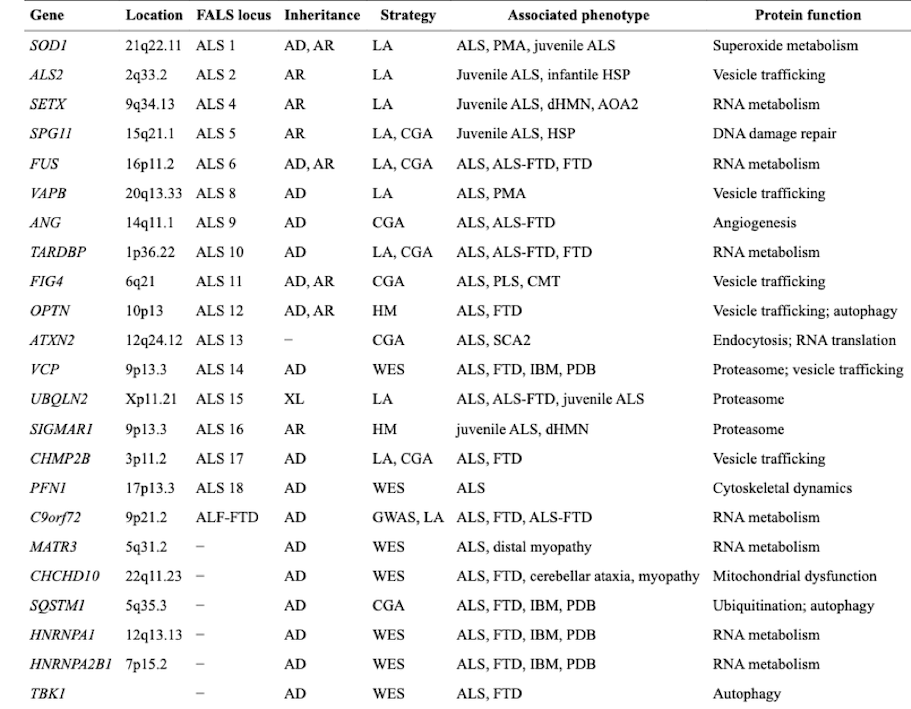
Table1: Summary of genes linked to Amyotrophic Lateral Sclerosis (ALS)[15].
Environmental Factors
Environmental factors play a role in the development of ALS where no family history of the disease is present, those represent around 90% of cases.The main causative factor may be head injury. A review conducted in 2015 found that moderate to severe traumatic brain injury is a risk factor for ALS, but whether mild traumatic brain injury increases rates is still unclear[11].
Several studies identified sports as soccer and american football to be risk factors of ALS. However, this study was based on small sample size of ALS cases[12]. Moreover,a 2011 meta-analysis concluded that smoking increases the risk of ALS. Among smokers, the younger they started smoking, the higher the risk of ALS was found to be. However, neither the duration of years of smoking nor the number of cigarettes smoked per day affected their risk of developing ALS[13].
References...