Article topic: Cerebral ischemia
Author: Areen Amaireh
Keywords: cerebral, infarction, ischemia, hypoxia, occlusion.
Definition and symptoms
Cerebrovascular diseases are brain disorders caused by pathological processes involving blood vessels (1). Ischemic cerebrovascular events represent a significant public health problem because of their high morbidity, mortality, and possible disability (2).
The three main pathogenic mechanisms are thrombotic occlusion, embolic occlusion, and vascular rupture. Stroke is the clinical designation applied to all these conditions when symptoms begin acutely. Thrombosis and embolism have corresponding impacts on the brain: loss of oxygen and metabolic substrates, ensuing in infarction or ischemic damage of regions supplied by the affected vessel. A similar injury occurs globally when there is a complete loss of perfusion, severe hypoxemia, or profound hypoglycemia. Hemorrhage accompanies rupture of vessels and causes immediate tissue damage, as well as secondary ischemic, injure (1).
The symptoms of cerebral ischemia depend on the anatomical region undergoing ischemia. Ischemia within the branches from the internal carotid artery may result in symptoms such as unilateral blindness, weakness in one arm or leg, or weakness in one entire side of the body. Ischemia within the branches from the vertebral arteries in the back of the brain may result in symptoms like dizziness, vertigo, double vision, or weakness on both sides of the body.
There are two kinds of ischemia: focal ischemia, represented by a particular region of the brain; and global ischemia which includes wide segments of the brain (3) figure (1): hemorrhagic stroke
figure (1): hemorrhagic stroke
Figure (2): ischemic stroke
 Global Cerebral Ischemia
Global Cerebral Ischemia
In the severe setting of systemic hypotension, usually, when systolic pressures fall below 50 mm Hg, widespread ischemic-hypoxic injury can occur. The morphology of global cerebral ischemia shows a swollen in the brain, with wide gyri and narrowed sulci. The cut surface exhibits poor demarcation amidst gray and white matter. Histopathologic changes that accompany irreversible ischemic injury are grouped into three categories: Inaugural changes, happening 12 to 24 hours after the injury, embrace acute neuronal cell change, characterized initially by micro vacuolation, followed by cytoplasmic eosinophilia, and finally nuclear pyknosis and karyorrhexis. Similar changes to astrocytes and oligodendroglia are somewhat witnessed later. Consequently, the reaction to tissue damage begins with neutrophil (PMN) infiltration. Subacute alterations, creation at 24 hours to 2 weeks, include necrosis of tissue, the influx of macrophages, vascular proliferation, and reactive gliosis. Repair, seen after 2 weeks, is characterized by the removal of necrotic tissue and gliosis (1).
 Figure (3): Global cerebral ischemia, Case courtesy of Dr Mohammed Al-Hindawi (4).
Figure (3): Global cerebral ischemia, Case courtesy of Dr Mohammed Al-Hindawi (4).
Focal Cerebral Ischemia
Focal ischemia is confined to a specific area of the brain (3). Cerebral arterial occlusion first leads to focal ischemia, and later to an infarction in the distribution of the compromised vessel. The size, location, and shape of the infarct and the extent of tissue damage that results may be compensated by collateral blood flow. Specifically, collateral flow through the circle of Willis can limit damage in some areas (1).
 Figure(4):focal cerebral ischemia (5).
Figure(4):focal cerebral ischemia (5).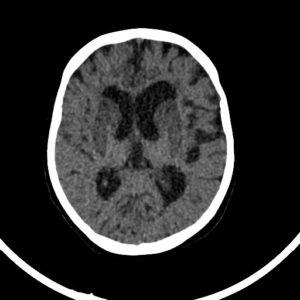 Figure (5): Hypoxic ischemic encephalopathy (HIE), Case courtesy of Dr. Abdallah Mohamed (6).
Figure (5): Hypoxic ischemic encephalopathy (HIE), Case courtesy of Dr. Abdallah Mohamed (6).
Etiology
Brain ischemia has been linked to a variety of disorders or abnormalities. People with sickle cell anemia, compressed blood vessels, ventricular tachycardia, plaque buildup in the arteries, blood clots, very poor blood pressure as a consequence of heart attack, and congenital heart defects have a higher exposure to brain ischemia in comparison to their healthy counterparts (3).
Due to thrombosis, embolic infarctions are more common than infarctions. Cardiac mural thrombi are a frequent source of emboli; myocardial dysfunction, valvular disease, and atrial fibrillation are important predisposing factors. Thromboembolic events also arise in arteries, most often from atheromatous plaques in the carotid arteries or aortic arch. Emboli of venous origin may cross over to the arterial circulation through a patent foramen in the brain, these include thrombo-emboli arising from deep leg veins and fat emboli, usually following long bone trauma like the femur. The territory of the middle cerebral artery, a direct extension of the internal carotid artery, is most frequently affected by embolic infarction. Emboli tend to lodge where vessels branch or in areas of stenosis, usually caused by atherosclerosis (1).
Pathophysiology
The brain is a greatly oxygen-dependent tissue that necessitates continual provisioning of glucose and oxygen. Despite it making up no more than 2% of body weight, the brain gets 15% of the resting cardiac output and is responsible for 20% of the entire body’s oxygen consumption. Cerebral blood flux normally stays stable over a wide range of blood pressure and intracranial pressure due to autoregulation of vascular resistance.
The brain may be deprived of oxygen in two ways:
- Functional hypoxia, caused by a low partial pressure of oxygen, weak oxygen-carrying capacity, or toxins that interfere with oxygen use.
- Ischemia, either transient or permanent, due to tissue hypoperfusion, which can be caused by hypotension or vascular obstruction (1).
Ischemic cascade refers to many interrelated and overlapped pathological mechanisms that are activated after a few minutes of blood vessel occlusion, and whose advance occurred a various times. The initial event of the ischemic cascade is the reduction of oxygen and glucose, causing failure in high-energy molecule production responsible for cellular homeostasis maintenance. This sets off several mechanisms that include ionic imbalance, excitotoxicity, calcium overload, cytotoxic and vasogenic edema, peri-infarct depolarization, oxidative and nitrative stress, blood-brain barrier disruption, inflammation, and apoptosis (7).
Pathogenesis
Within seconds of cerebral ischemia, local cortical activity as discovered by electroencephalography ceases; if the ischemia is global, unconsciousness speedily ensues. This massive shutdown of neural activity is induced by K+ efflux from neurons, mediated initially by the opening of voltage-dependent K+ channels and later by ATP-dependent K+ channels, resulting in transient plasma membrane hyperpolarization. A few minutes later, despite this energy sparing response, a severe and dramatic redistribution of ions occurs across the plasma membrane, associated with membrane depolarization (efflux of K+ and influx of Na+, Cl–, and Ca2+). This “anoxic depolarization” results in the excessive release of neurotransmitters especially glutamate, promoting a more spatial spread of cellular depolarization, depletion of energy stores, and advancement of harm cascades (8).
Obviously, lack of energy causes initially an electrical failure. If energy depletion lasts longer, the arrest of cellular functions and cell death takes place. However, animal trials and clinical studies display that there are other factors, like energy loss, that account for neuronal damage. Even sublethal hypoxic-ischemic encephalopathy can set in motion a series of toxic reactions that finish off injured neurons and kill additional ones that have not been damaged during the initial injury. Accordingly, following global ischemia, neurons do not die suddenly or all at once. In some cases, global ischemia leads to bilateral, symmetric cerebral infarcts in the border zones amid major arterial territories. Rarely, hypoxic-ischemic encephalopathy involves the white matter, causing myelin damage and loss. Dismantling of damaged myelin results in vacuolization and a spongy appearance of the white matter in tissue parts. White matter damage is common in carbon monoxide (CO) poisoning but may occur in other forms of hypoxic-ischemic encephalopathy (9).