Article topic: Transverse Myelitis.
Author: Rua’a Nader Abd Al-Qader.
Editors: Nicola Al-Madani, Ethar Hazaimeh
Keywords: Transverse Myelitis, Immune-mediated myelopathy, inflammatory, demyelinating, neurology, spinal cord.
Overview and epidemiology
Myelopathy is a broad, generic condition that does not have a particular etiology, whereas myelitis is an inflammatory disease, and both of them affect the spinal cord [1].
Transverse Myelitis (TM) is characterized by acute or subacute spinal cord dysfunction, leading to paresis, decreased sensations, or autonomic impairment (bladder, bowel, or sexual) below the level of the lesion [2][3]. It is considered as inflammation across the entire transverse section of the spinal cord, which is called complete transverse myelitis, and there are many cases referred to as partial transverse myelitis, the clinical presentation of these two conditions can overlap [4]. This inflammation results from infectious, immune-mediated, or non-inflammatory myelopathies, such as compressive, vascular, neoplastic, metabolic, nutritional, toxic, and inherited causes [5].
Transverse myelitis is classified into disease-associated that is related to infectious or immune-mediated etiology, and idiopathic cause, which should be a diagnosis of exclusion [4]. That’s made great progress in the autoimmune neurology field for discovering neural autoantibodies (eg, AQP4-IgG and myelin oligodendrocyte glycoprotein IgG), and this helped to know a specific cause for patients previously diagnosed with Idiopathic TM [6][7].
When we talk about the epidemiology of this disease, we should focus on race when evaluating patients with immune-mediated myelopathies. Neurosarcoidosis is common in African Americans [8], Multiple Sclerosis (MS) is numerous in whites and regions away from the equator, whereas the incidence of idiopathic transverse myelitis in 2019 was 8.6 per million, and the prevalence was 7.9 per 100,000 [7]. In general, the annual incidence of TM ranges from 1.34 to 4.60 cases per million [9][10][11], but increases to 24.6 cases per million with demyelinating diseases like MS [12].
Pathogenesis and etiology
1- Acquired demyelinating disorders
a. Multiple Sclerosis (MS)
MS is a progressive, demyelinating neurologic illness characterized by an immune-mediated inflammatory response. The first attack of MS, which is called Clinically Isolated Syndrome (CIS), might present with acute optic neuritis (AON), or acute partial transverse myelitis (APTM). The chance of APTM developing into Clinically Defined Multiple Sclerosis (CDMS) varies between 10% and 62 percent [13]. The most prevalent symptom of TM in MS is sensory impairment.
The MRI is the most crucial test for determining the likelihood of transitioning to CDMS. Only 10% of individuals with APTM will develop CDMS at 61 months if it is normal [13], but this increases to a 21% risk of progression to CDMS at 20 years follow-up, according to another study [14].
Another important factor that predicts the risk of progression to CDMS is CSF Oligoclonal bands (OCBs). In the case of TM, OCBs have been revealed a predict conversion to MS [15]. The presence of OCBs and/or an increased IgG in individuals with TM who have normal MRIs indicates a greater risk of developing MS [16]. With a normal brain MRI and CSF results, the probability of transitioning to MS is less than 10% [15].
b. Neuromyelitis Optica (NMO)
Patients with NMO usually coexist with autoimmune diseases, such as Systemic Sclerosis (SS), Systemic Lupus Erythematosus (SLE), Autoimmune Thyroid Disease, Type 1 Diabetes Mellitus, Ulcerative Colitis (UC), and Myasthenia Gravis [17][18]. All patients with known autoimmune diseases, who present with TM, undergo serologic testing for NMO-IgG. The high specificity of positive NMO-IgG will work up the additional diagnosis of NMO coexisting with the systemic autoimmune disorder [19][20].
c. Acute Disseminated Encephalomyelitis (ADEM)
ADEM is a monophasic disease that happens following infections or vaccinations. It is linked with multifocal demyelinating lesions and may compromise encephalopathy, coma, or seizures [21]. TM takes place in about 24% of patients [22]. Unlike MS and NMO, ADEM is possibly associated with demyelinating peripheral neuropathies [23]. ADEM is likely to occur at any age, but most commonly, in pediatric patients (mean age is 5.7 years) [24]. Post-vaccination ADEM incidence is changeable, and the most frequently involved vaccine is the non-neural measles, mumps, and rubella vaccine [25][26].
Typically, the spinal cord lesion is swollen, and mostly affects the thoracic cord [22]. CSF in ADEM shows marked pleocytosis, raised protein, normal IgG index, and no Oligoclonal bands (OCBs) [26].
2- Systemic inflammatory autoimmune disorders
a. Sjogren Syndrome (SS)
SS is a chronic, progressive, inflammatory autoimmune disease that occurs mainly in middle-aged to elderly women, as the immune system mainly attacks lacrimal and salivary glands, leading to keratoconjunctivitis sicca or sicca syndrome. SS may occur alone (primary SS) or exist with underlying autoimmune disease (secondary SS)[27][28][29].
A wide range of CNS symptoms may occur, including acute optic neuritis (AON) and transverse myelitis (TM) [28][30][31]. Neurological manifestation may be the initial presentation in 57% of patients with SS [29]. Involvement of spinal cord (either acute TM or progressive myelopathy) possibly happens in 20% to 35% of patients with SS and may compromise the initial presentation of the disease in up to 20% [27][29]. The lesions affect the cervical cord and may extend longitudinally [29][32].
Typically, CSF results in pleocytosis, mildly elevated protein, and a mildly increased IgG index [28][32]. Careful follow-up is important in SS, as recurrent attacks of TM or AON can end in substantial disability, and may lead to a confirmed diagnosis of NMO or MS[1].
b. Systemic Lupus Erythematosus (SLE)
SLE is a chronic multisystem autoimmune disease. The most ruining complication of SLE is TM, which accounts for only 1% to 2% of patients [33], and it is considered to have a poor prognosis[25].
SLE-related TM occurs within the first 5 years from diagnosis. TM is the initial neurological manifestation in half of the patients and recurs in 21% to 55% of cases [34][35][36]. AON and brainstem manifestations may go along with TM in SLE [37].
In many cases of SLE-related TM, two different clinical patterns may patients present with, gray or white matter myelitis. Gray matter myelitis presents with lower motor neuron (LMN) manifestations, urinary retention, and it happens in a monophasic, devastating course. White matter myelitis presents with UMN manifestations, it occurs in more indolent and recurrent courses [38].
c. Behcet Disease (BD)
BD is a relapsing systemic inflammatory disease of unclear cause, which presents with oral aphthous ulcers, genital ulcers, uveitis, cutaneous symptoms, and involvement of other systems[39].
Neurologic participation in BD (neuro-BD) occurs in the 3rd to 4th decade of life, and it is more common in men, often associated with ocular involvement [40][41][42].
Spinal cord involvement ranges between 2.5% and 30.0%, mainly the cervical and thoracic cord segments (specifically the posterolateral cord), and has a poor prognosis [40][41]. TM in neuro-BD alone is unusual [40][43]. Spinal cord lesions are longitudinally extensive and include multiple non-contiguous segments, or even the whole cord [44][45].
CSF shows normal glucose, elevated protein, and neutrophilic pleocytosis; IgG index may be increased, and OCBs are rare[41][46].
3- Parainfectious TM (PITM)
Parainfectious TM refers to TM associated with previous infection. It is hard to determine if PITM is caused by direct microbial invasion causing myelitis or a consequence of immune-mediated, inflammatory reaction induced by a distant infection. The previous infection has resolved before the onset of TM and it is difficult to illustrate the offending organism in the spinal cord parenchyma [1].
The hepatitis viruses possibly cause TM by post-infectious, immune-mediated, inflammatory reaction[47]. Hepatitis A virus and Hepatitis B virus infection are associated with immune-mediated TM [47]. Hepatitis C virus (HCV) is the most commonly involved hepatitis virus in TM. HCV referred to recurrent, corticosteroid-responsive, demyelinating TM (even without hepatic association)[47][48]. There is a single report of hepatitis E–virus related to TM[49].
Regarding Mycoplasma Pneumoniae infection and its neurological symptoms that are considered the most common extra-pulmonary complication of this infection [26][50]. It was found that encephalitis is the most frequent complication [51], and TM is the most severe and serious manifestation[52]. Acute to subacute thoracic myelopathy usually appear 2 to 4 weeks after a previous respiratory infection and progresses to a severe condition in about 3 days; it may be associated with meningoencephalitis and/or polyradiculopathy [52].
These microbes most likely cause myelopathy by the direct pathogenic attack. A careful follow-up is needed and more evaluation is to be held, because infections may trigger TM that precedes the diagnosis of MS or NMO[1].
4- Paraneoplastic TM
Collapsin response-mediator protein-5 (CRMP-5-IgG) antibody that has been appeared in small cell lung cancer, is the most commonly paraneoplastic antibody associated with TM [53][54].
Patients present with subacute, progressive motor myelopathy mainly as well as elevated CSF protein, increased IgG index, and mild pleocytosis [53][55].
5- Atopic Myelitis (AM)
Atopic myelitis (AM) presents with the chronic, persistent, or fluctuating course of cervical myelitis mainly. It is associated with hyper-IgEemia, allergen-specific IgE (due to dust mites), and coexistent atopic diseases (eg, atopic dermatitis, atopic rhinitis, or asthma) [56].
The majority of reported cases happen in Japanese patients, but there are few cases of AM in White patients[57]. Sensory impairment is a predominant symptom, with uncommon motor and bladder involvement[58]. CSF cell count and protein are mostly normal and OCBs are not seen[59].
6- Drug-induced and Toxin-related TM
– TNF-alpha inhibitors cause CNS demyelination and TM [60].
– Sulfasalazine [61].
– Chemotherapeutic agents: Gemcitabine, cytarabine, and cisplatin [62].
– Heroin: most of the cases of myelopathy in heroin addicts are related to anterior spinal artery infarction, moreover, there are cases of TM[63].
– Benzene [64].
– Brown recluse spider bite: Incomplete TM (anterior spinal syndrome), responds to steroid therapy[65].
7- Idiopathic TM (ITM)
The proportion of patients with ITM differs widely, from 16% [66] to 60%[67]. The mean age of onset occurs between 35 and 40 years, mainly in females [66].
CSF shows elevated protein in most patients; pleocytosis and OCBs are seen occasionally [66][68]. Unlike other causes of TM, negative OCBs are related to recurrence [66].
ITM is monophasic, but it recurs in about 1/4 to 1/3 of patients (recurrence of primary insult, enlargement of prior lesion, or a new lesion) [66][69].
Risk factors for recurrence are male gender, age older than 50 years, severe motor weakness and sphincter dysfunction and negative CSF OCBs, normal IgG, and seronegative NMO-IgG [66][68][69]. If recurrences happen, they will lead to poor outcomes [69].
Generally, one-third of patients with ITM recover with little or no consequences, one-third have a moderate extent of permanent disability, and one-third have severe disabilities[66].
Clinical presentation and complications
Transverse Myelitis has an acute or subacute onset; with neurological dysfunction extending to very severe manifestations within a few weeks, and it may present as one of many syndromes of the spinal cord[1].
The typical clinical symptom of myelitis is to have sensorimotor deficits in one or more extremities. Sensory impairment, like ascending numbness is a characteristic presentation of TM. Some patients present with a rounded band of dysesthesia attributed to the dermatomes just rostral to the sensory level around their trunk, and this may be associated with constricting sensation (as the “MS hug”) that ranges from mild discomfort to severe burning pain [1]. The pain associated with TM can be central, deep aching, or radicular pain [3].
Autonomic dysfunction is common, and it presents as the neurogenic bladder (urinary retention), neurogenic bowel, or sexual dysfunction; these symptoms are common with Myelin Oligodendrocyte Glycoprotein IgG (MOG-IgG)–associated disorder, also maybe reflect conus involvement radiologically [4]. Autonomic impairment also could be presented with temperature dysregulation or even bouts of hypertension[11][70].
Motor symptoms usually show weakness that can affect the flexors of the legs and extensors of the arms (pyramidal distribution) or can present with sphincter dysfunction [70]. Acutely, muscle tone and stretch reflexes could be reduced or absent, which is called “spinal shock syndrome” and that makes diagnostic confusion with Guillain-Barre syndrome (GBS). With time, spasticity, hyperreflexia, and extensor plantar responses (ie, features of the upper motor neuron lesion) become apparent[1].
Clinical manifestations of a “demyelinating cause” involve the Lhermitte sign (an electrical sensation that spreads down the spine and extremities when the neck is flexed), and the Uhthoff sign (transient worsening of neurologic symptoms when the body is overheated). Also, tonic spasms (recurrent short events of involuntary painful contractions by flexors that last 30 seconds to a few minutes) may present [4].
In addition, we should ask about symptoms that are related to systemic autoimmune diseases (like oral or genital ulcers in Behçet disease) or sicca symptoms, also maybe the presence of arthritis or skin changes. Moreover, we should look for symptoms that suggest myelopathy related to connective tissue diseases [4].
Evaluation and diagnosis
MRI of the whole spinal cord is obligatory in any patient with myelopathic manifestations, in order to exclude structural lesions that are responsive for emergent neurosurgical intervention. The most sensitive MRI sequence to identify spinal cord lesions (particularly MS plaques) are short-tau inversion recovery (STIR) fast spin-echo and T2-weighted fast spin-echo[71][72]. The location and length of the lesion on MRI could give signs of the underlying disease (Fig 1), (Fig 2).

Fig 1. Extensive increased T2 signal and expansion of the cord is seen extending between C7 and T12. The T2 signal abnormality involves central grey matter and dorsal columns [85].
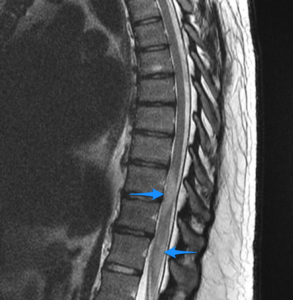
Fig 2. increased T2 signal and mild swelling (blue arrows) involves most of the conus, more patchy towards the tip [86].
Vitamin B12 levels, thyroid function tests, syphilis, and HIV serology are acquired for evaluation of treatable causes of myelopathy. Vitamin E, serum copper, and ceruloplasmin levels should be checked in patients at risk of deficiency. Serum aquaporin-4–specific autoantibodies (NMO-IgG) should be investigated on all patients with TM due to high specificity for NMO [19][74].
Inflammatory markers should be assessed if systemic autoimmune diseases are suspected. In suspected Parainfectious TM, serologic titers of recent infection should be requested. Paraneoplastic profiles should be done in cases of paraneoplastic TM; moreover, in these cases, proper investigations should be made to look for hidden malignancy[1].
Cerebrospinal fluid (CSF) analysis is necessary for the evaluation of TM. CSF cell count, protein, glucose, oligoclonal bands (OCBs), and IgG index should be assessed on all patients with TM [75]. OCBs are useful in portending transition to MS, because OCBs are present in 85% to 90% of patients with MS, in 20% to 30% with NMO, and rarely in other etiologies of TM [73].
A neuro-ophthalmological evaluation is required to look for ophthalmic symptoms that could give us important diagnostic clues, especially when radiologic and lab investigations are unclear [76]. Demyelinating diseases that attack the brainstem or cerebellum usually cause ocular motor manifestations[77].
Electro-physiologic tests also could be useful in evaluating patients with TM. Nerve conduction studies (NCS) and electromyography (EMG) could help to show any peripheral nervous pathology[1].
Treatment
For most autoimmune myelopathies, after exclusion of other etiologies such as compressive lesions, spinal cord infarction, and infections, it is recommended to administer high-dose IV corticosteroids with 1 g IV methylprednisolone once daily for 5 days duration [4].
In patients with myelitis as a symptom of CNS demyelinating disease (eg, MS, MOG-IgG–associated disorder, ADEM, idiopathic transverse myelitis) with severe neurological manifestations, plasma exchange (plasmapheresis) should be considered [78][79].
Early starting of plasmapheresis (within 15 days of onset) gives a favorable acute response and improvement at 6 months [80]. Typically, the regimen is to exchange 1.5 plasma volumes for five treatments within 10 days [81].
In children with MOG-IgG–an associated disorder of TM, IV immunoglobulin (IVIg) is used acutely, and it was shown an improvement of the outcome, also using steroids over 6 to 12 weeks could be considered after an acute attack to prevent early relapse [4].
Supportive care should be considered to prevent complications; it could be an implementation of bladder scans or temporary catheter placement, and prevention of deep venous thrombosis and ulcers. Depending on the cause of the TM, chronic immunomodulatory therapies will help to prevent future relapses and attacks [4].
Prognosis
The prognosis of acute transverse myelitis predicts a poor outcome, involving rapid progression of neurological manifestations, back pain, spinal shock, and absent central somatosensory conduction [66], but early initiation of plasma exchange has a better outcome [80]. Some cases revealed that 1/3 of patients recover with a few or no lasting symptoms, another 1/3 have a moderate disability, and 1/3 have a severe disability [82]. Furthermore, the long-term prognosis is highly influenced by determining the underlying etiology of transverse myelitis [83].
Does COVID-19 cause Transverse Myelitis?
Recently, it was shown an association between COVID-19 infection and the development of different types of CNS demyelination such as Transverse Myelitis. Some patients had an isolated TM and others were part of diffuse demyelinating disorders. Longitudinal Extensive Transverse Myelitis (LETM) was the most frequently reported of spinal involvement in 72.5% of cases. There are several mechanisms by which SARS-CoV-2 can cause myelitis: acute viral myelitis, post-COVID-19 immune-mediated myelitis, and inflammatory demyelinating disorders that could be triggered by COVID-19 infection (ADEM, MS, and NMO). However, this association should be clarified in future literature [84].