Title: Central Nervous System Primitive Neuroectodermal Tumor.
Author: Ghadi Ahmad Hasan Al-Wazani.
Keywords: Oncology, Neurosurgery, Tumor classification, PNETs.
Scientific Editor: Dr Omar Jbarah
Introduction :
The terminology Central Nervous System Primary Neuroectodermal Tumors (PNET) have been removed from the diagnostic dictionary1 and was replaced by new tumor names that reflect the new classification of these tumors. This new classification is based on newly discovered genetic and molecular classification.2 Before the C19MC gene amplification discovery, all tumors that were not Medulloblastoma were classified as CNS Primary Neuroectodermal Tumors (PNET) , or supratentorial PNETs. Nonetheless, they are now classified into different categories depending on molecular differentiation.3
In this new classification (WHO 2016 classification), supratentorial High grade Neuroepithelial Tumor with BCOR alteration, for example, were discovered to be high grade Gliomas or Ependymomas that were misdiagnosed as Primary Neuroectodermal Tumors (PNET) before the 2016 WHO CNS PNETs’ new classification. The misdiagnosis was present in 71% of the cases.4
Other subtypes include Cerebral Neuroblastoma and Ganglioglioneuroblastoma. The identification of each of the previous two subtypes depend on the ganglionic differentiation. 5
In general, these tumors are aggressive, poorly differentiated, or undifferentiated, and arise from embryonal cells. They appear in children in most cases but may present in few cases in adolescents and adults. 2
The WHO most recent classification divides them into CNS Medulloblastoma, Embryonal Tumors with Multilayered Rosettes/ C19MC altered, CNS Neuroblastoma, and Atypical Teratoid/ Rhabdoid Tumors. Pineoblastoma have been removed from the group due to its unique molecular characteristics. 3
When these tumors were compared through radiological imaging, they were found to be similar in their edematous changes, definition of margins, and size. Therefore, they are radiologically identical. 4
Immunohistochemical staining demonstrated the unique characteristics for each tumor group.4 Important molecular markers include Sonic Hedgehog (SHH) and Wingless-related Integration Site (WNT). SHH is a protein that functions in cell specialization and growth and is encoded on the 7th chromosome, while WNT is encoded on chromosome 12 and is responsible for cell proliferation, cell fate and embryogenesis. Both markers are important in Medulloblastoma diagnosis and classification. 2
Medulloblastoma (MDB):
It is considered the most common among embryonal tumor and is deemed WHO grade 4. They reside in the cerebellum, either in the vermis or in the cerebellar hemispheres depending on the age of occurrence. In children, they arise in the vermis and in adults in the hemispheres. In addition, the have two peak ages. Those are 7 years and 21-40 years.5
MDBs are frequently diagnosed after the appearance of clinical symptoms. 6
Their histology shows sheets of densely packed cells with scarce cytoplasm and a large round nucleus. The mitotic figures are plentiful, and neuroblastic Rosettes (Homer Wright) that represent 40% or less of the tumor. It is uncommon for this tumor to have vascular proliferation with Pseudopalisading Necrosis. Their gross appearance is relatively well-circumscribed but has Leptomeningeal involvement in one third of the patients. 5
Medulloblastoma has different variants that are based on their morphology. In Nodular or Desmoplastic Variant, the surface of the tumor appears nodular like a lymph node. Within them is poorly differentiated cells that produce reticulin-positive collagen fibers in excess amount intercellularly, and islands that are reticulin free. This variant appears in the cerebellar hemispheres 5 1, and midline.1 They present bimodally in young children (Gorlin positive) + adolescents, and in adults.1 In addition, it has better prognosis than other variants.5
Another variant is the rare infantile variant, which is the Densely Nodular Variant.5 In <50% the appear in children <3 years old.1 Its features include having many neurocystic nodules.1 Microscopically, they have large lobules, with neuropil-like tissue, neoplastic neurocytes, and few or non-extracellular tissue.5 They are in the vermis in 80% of the cases.1 They have high mitotic activity, pleomorphism, prominent nuclei, and high rate of necrosis.5
The Classic Variant have densely packed small round undifferentiated cells with high count of mitosis, and few pleomorphism. They present in 72% of the cases and can arise in all ages but predominantly in childhood.1
The Anaplastic Variant, which has pleomorphic nuclei with cell-cell wrapping, and nuclear molding.5
The last variant is the Large Cell Variant. This variant has cells that shows more monomorphism and are with a large-vesicular-rounded nucleus. The last two variant have morphologies that overlap, high mitotic rate, high apoptotic rate, and are both aggressive.5
Medulloblastoma is divided into different groups as well depending on its positivity to neuronal markers. All MDBs have glial differentiation that makes them Glial Fibrillary Acidic Protein positive in bona fide neoplastic cells. In Medulomyoblastoma, there is myogenic differentiation (Desmin, Myosin, and Myoglobin positive), while in Melanocytic MDB it has melanotic differentiation.5
MDBs are divided into clusters these are:
1) WNT- activated 10% of MDBs:1 they represent 10% of MDBs. This cluster occurs in older children and rarely in infancy. In addition, it has female to male predominance of 2:1. Their cell of origin is the lower rhombic lip progenitor cells. Their histology shows mostly the classic variant (low risk), while rarely the Anaplastic or the Large Cell variants. They favor the midline.2
2) SHH activated/ TP53 wild type: they have no gender predominance and occur in children and adults. Their cell of origin is the cerebellar granule neuron cell precursor in external granule layer and in cochlear nucleus. The predominant histology is the Desmoplastic/ Nodular variant (low risk). In infants, they have the Extensive Nodularity subtype predominantly (low risk). Other morphologies include the classic morphology (low risk), and the Large Cell morphology (uncertain clinical significance).1
3) SHH activated, TP53 mutant: it is present in childhood typically with no gender predominance. It has the same cell of origin as the TP53 wild type. The morphologies: Large Cell (high risk) is prevalent in patients aged 7-17 years, Desmoplastic/Nodular very rarely, and Classic morphology is uncommon (high risk). TP53 mutant and TP53 wild type represent 30% of the cases.1
4) None SHH, none WNT Group 3: it arises from the same cells as the previous two groups but has a male to female predominance of 2:1. The histology includes Classic variant (standard risk), and the Large variant (high risk).1
5) None SHH, none WNT Group 4: it is present in all ages group with male to female predominance of 3:1. Their cell type is unknown, and have their morphological variants are the Classic Variant (standard risk) and rarely Large/ Anaplastic variant.1
6) MDB/ NOS: has no genetic or histologic classification. This cluster must be differentiated from none MDB tumors before making the diagnosis. Including high grade small cell glioma, embryonal tumors that are with multilayered rosettes, atypical/rhabdoid tumors, and tumors in either the 4th ventricle or the cerebellum.1
Latest publications have, in fact, identified up to 12 subgroups for MDBs.
MDBs are the most common pediatric age tumors, which occur in the first decade of life.1 They represent, as well, 2% of CNS tumors in adults.7 They are located usually in the apex of the vermis (fastigium), in the roof of the fourth ventricle, in the posterior aspect of the brain stem, or, rarely, they can be supratentorial.1
MDB prognosis is dependent on multiple factors, with the following being responsible for poor prognosis: the age of the patient (1.5 cm2 postsurgical residual tumor), the presence of leptomeningeal spread, and poor Karnofsky performance score.1 In 30% of patients, MDBs will present with leptomeningeal dissemination. However, it is rare that these tumors present with metastasis outside CNS. The five-year overall survival is 70%.8
Recurrence, when it happens, is in the posterior fossa.
MDB can be divided into three risk groups: standard risk, low risk, and high risk.
1) Standard risk: when the patient has total or near total resection (1.5 cm2 on early postsurgical axial plane MRI. They include patients as well with extra-CNS metastasis, or in tumors with Large Cell/ Anaplastic histology in group 3 or SHH activated with TP53 mutation.1
This risk stratification is determined to guide the management of the patient. However, this is not the only system that is present in classifying MDBs. The near total resection criterion is a controversial one, since it was placed during CT-era.1
Another staging system is Chang Staging System (the most MDB robust staging system), and is described below:
M0= no signs of gross subarachnoid metastasis.
M1= microscopic cells are found in the CSF.
M2= it has gross nodular seeding in either the cerebellum, cerebellar subarachnoid space, in the 3rd ventricle, or in the lateral ventricles.
M3= the seeding is in the subarachnoid space of the spine, or if there is a gross nodule.
M4= the tumor has extraneural metastasis. 1
*Embryonal Tumors with Multilayered Rosettes:
These tumors have true Rosettes that are over a background of neuropil. It is both rare and aggressive tumor.5 They affect children below the age of four years. 9 10, with the majority being below two years old. They are in the cerebrum, brain stem, the cerebellum,5 3 Pineal Gland, and rarely in the orbits.3 Their genetic variation is having an amplification mutation of C19MC gene on chromosome 19. Nonetheless, when they lack this mutation, they are called ETMR, NOS. both variants are WHO grade 4.1
Three subtypes have been identified. These are Embryonal Tumors with Abundant Neuropil and True Rosettes, Ependymoblastoma, and Medulloepithelioma.9 10The C19MC amplification was demonstrated in 93% of all subtypes. The other very well-expressed protein, and which is specific and sensitive for ETMR subtypes, is LIN28A. The detection of this protein is a dependable and a fast diagnostic method. Nonetheless, in AT/RT LIN28A is expressed but in a lower percentage. In centers where molecular testing is not possible, the histological appearance of multilayered rosettes is obligatory for diagnosis. 9
Histologically they have small round blue cells with a pseudostratified neuroepithelium that surrounds a lumen that is either empty or has eosinophilic waste material. They are positive for Cytokeatin, CD99, and Vimetin. While being focaly positive for Epithelial Membrane Antigen. In addition, their Ki-67 labeling index is remarkably high.2
ETMR reflects the histological appearance present in most, but not all C19MC altered tumors. 3
Ependymoblastoma, which lack neuropil on histology, Medulloepithelioma, and Embryonal Tumor with Abundant Neuropil and True Rosettes are all put under the classification of EMTR in the WHO 2016 classification. Medulloepithelioma exhibits papillary and tubular structures that are like neuronal tube with variable neuropil. In 20-25%, ETMR will have no Rosettes, no Neuropil, but has inconstant differentiation with probable intratumor heterogenicity of the cells. Due totheir heterogenicity and rarity, advances in the methods of diagnosis for ETMR is vital. 3
C19MC altered tumors are in about 65% of the cases localized at diagnosis. Nonetheless, they constantly have rapid prognosis and are resistant to treatment. 3
")
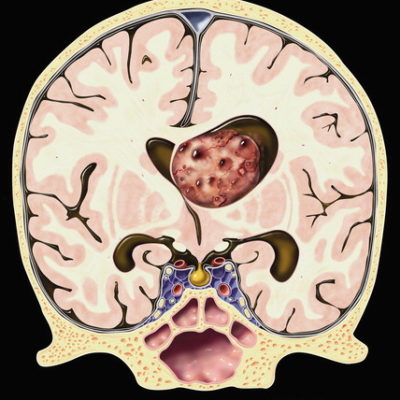
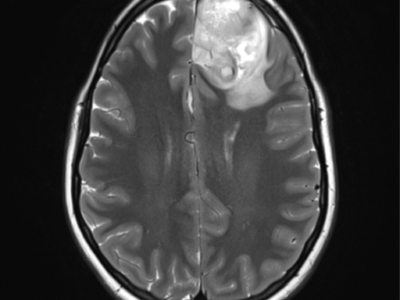
Figure 1. Case courtesy of Dr Antonio Rodrigues de Aguiar Neto Radiopedia.org 11
*Atypical Teratoid/ Rhabdoid tumors (AT/RT):
AT/RT are tumors that are rare.5 By definition, they are poorly differentiated cells primitive neuroectodermal cells with rhabdoid cells.1 In 75% of the cases they will be present in the posterior fossa with the rest being supratentorial. 5 Infratentorial location occurs in the Cerebellar hemispheres, Cerebropontine angle, and brain stem. 1 This tumor is a tumor of childhood and can have neuronal, glial, epithelial, mesenchymal, or a mixture of these differentiations. 5
The genetic mutation they express is SMARCB1 (INI1), or, extremely rarely, SMARCA4 (BRG1).1
Histologically, the tumor has pleomorphic cells with few rhabdoid cells (an eccentric nucleus in an eosinophilic cytoplasm). Though, the diagnosis is made when loss of INI1 nuclear staining is lost due to gene mutation. 5
When the tumor has the histological distinction, but lacks the genetic differentiation, they are called CNS embryonal tumors with rhabdoid features.1 Few of these patients have primary renal RH tumor. 1
Pure Rhabdoid tumors outside the CNS differs from AT/RT in the absence of INI1 germline mutation, which is present in the AT/RT. When compared with MDB, MDB kept the INI1 gene expression as well. 5 This distinction is important since it is frequently misdiagnosed as MDBs.1 AT/RT tumors can be positive for Vimetin and EMA. 2
These tumors have poor diagnosis, but they have, nonetheless, different behaviors. In 33% of the patients at presentation they present with tumor spread. They are WHO grade 4.1
AT/RT tumors is divided into three groups:
1-The AT/RT-TYR: they occur in very young children and they are infratentorial.
2-The AT/RT- MYC: the subtype that presents in older children and they are usually supratentorial.
3-The AT/RT- SHH: in this subtype, the tumor occurs in both age groups.2
")
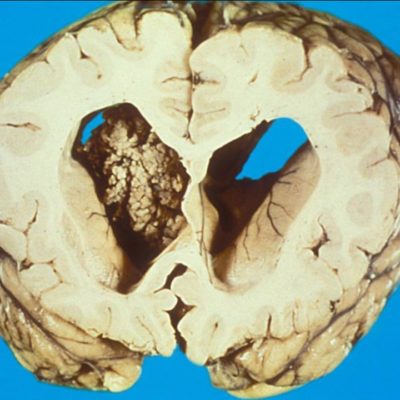
Figure 2. Case courtesy of Dr Bahman Rasuli. Radiopedia.org 12
*Neuroblastoma:
A very rare tumor that is poorly differentiated with neuroepithelial cells that resemble embryonic neuronal tube. 1 Their cells are small with hyperchromatic nuclei, surrounded by a halo that is clear, areas of neuropil with neurocyte or ganglion cells. Furthermore, they have nuclear palisades, vascular pseudorosettes, and Homer Wright Rosettes. Possibly, they could have microvascular proliferation. 2
Genetically, the majority are positive for FOXR1 gene mutation, 2 but some show FOXR2 gene fusion.3 They present in <4-year-olds with slight female predominance, and they are in the Cerebral Hemispheres. 3
CNS high grade neuroblastoma with MN1 mutation can have a combination of solid patterns with pseudopapillary ones that are associated with unique hyalinization, and they are GFAP positive. 2
They are classified as WHO grade 4. 1
")
Figure 3. Case courtesy of Journal of Neurosurgery. 13
* CNS Ewing Sarcoma Family with CIC alteration:
A highly cellular tumor with small round cells that are arranged in both alveolar and fascicular pattern. 2 The genetic alteration in CNS Ewing Family of Tumors is CIC gene fusion. This mutation, along with CNS high-grade Neuroepithelial Tumor with BCOR alteration, are present in extracranial soft tissue sarcomas that are poorly differentiated.3
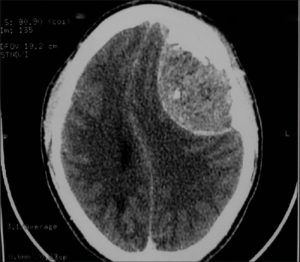
Figure 4. Case courtesy of Asian Journal of Neurosurgery. 14
*High grade Neuroepethelial tumors with BCOR alteration:
It is an extremely rare tumor that has a four-year survival of 50%.2 Their location can either be in the Cerebrum or the Cerebellum. Furthermore, it presents in <4-year-olds with equal male to female ratio. 3
The histological diagnosis of Astroblastoma is compromised by the M1N1 Neuroepithelial subtype. 3

Figure 5. Case courtesy of Springer. 15
*Emberyonal tumors, NOS:
These tumors must be differentiated from supratentorial Ependymoma. Their diagnosis is done through exclusion. Depending on genetic variation, they must be differentiated from high grade Glioma, ETMRs, and AT/RT. This group is the waste basket for tumors that were in the past categorized as CNS PNET and do not fit in any of the new molecularly based categories. 2
Clinical presentation
Patients with CNS tumors have presentations that depend on the location of the tumor (supratentorial vs. infratentorial) 1, age of the patient, and the type of tumor.4
Common presenting symptoms:
1) In 68% of the patients they will present with a progressive neurological deficit with 45% of them being a motor deficit.
2) In 54% the presenting symptom is headache.
3) In 25% they will have seizures that start as focal and progress to generalized. 1
Headaches are usually morning headaches that are thought to be due to hypoventilation during sleep. These headaches are exacerbated by coughing or straining, and in 30% while bending forward. Nausea and vomiting will be associated with headaches in 40% of the patients. In addition, the relieving factor for these headaches is vomiting. The presence of these characters with/without a focal neurological deficit and seizures is the description of classic tumor headache. Nonetheless, tumors present with a headache that is in 77% of the cases with a history that is Tension Headache like, in 9% Migraine like, and in only 8% the classic tumor headache presentation. 2/3 of the headaches come with an increase in intercranial pressure. 1
Supratentorial tumors present with increased intercranial pressure (ICP) due to direct mass effect of the tumor, edema or, less commonly, due to edema. They also present with progressive focal neurological defect, headache, seizure (not infrequent first visit symptom), mental status changes, and TIA symptoms. Transient Ischemic Stroke occurs due to occlusion of a blood vessel by tumor cells, or hemorrhage into tumor. 1
However, tumors that are in infant age group usually evade the diagnosis until the tumor reaches an enormous size. This is due to the expansile nature of the young child’s skull, the ability of their brain to recompensate for the lost function, and the difficulty in examining a patient with limited neurological range and is uncooperative. 1
The common symptoms of pediatric age group include a decline in psychomotor development, poor feeding, vomiting, macrocrania, ataxia, obstructive hydrocephalus (early if posterior fossa tumor), and seizures. The development of these symptoms usually occurs over a short period of time that is between 6 to 12 weeks. In the condition where there is spinal metastasis, there could be urinary retention, weakness, and back pain.1
Other possible symptoms include extraocular muscle palsies, truncal or appendicular ataxia, papilledema, and nystagmus.1
The most common symptoms in pediatric CNS tumors are headache in 37.2%, vomiting 33.3%, ataxia (most common symptom in older children), and oculo-visual symptoms.1
Variation in symptoms occur due to age as well. Nonetheless, symptoms that are associated with increased ICP arise in all age groups and all intercranial tumors.4 In younger children the presentation is of irritability, vomiting, visual disturbance,2 and nonspecific symptoms.4 Children older than 3 years, they will come with vomiting, ataxia, and headache.2 Cerebellar and brain stem signs are the most common presentation in Medulloblastoma. 7
The interval between initial clinical presentation and the diagnosis is short, estimated to be 20 days. The pre-diagnosis interval is even shorter in infratentorial tumors, high grade tumors, and younger age groups. 2 Medulloblastoma has a shorter pre-diagnosis interval of 15 days.4
The diagnosis must be tissue diagnosis.7
Investigations
In a CT scan without contrast, the tumor is hyperdense as a result to their high cellularity, while a CT scan with contrast will show an enhanced tumor, mostly, with calcification in 20% of the cases.1
In MRI, there will be a large, demarcated mass with edema surrounding it, and a significant mass effect. 2 T1W1 the tumor will be hypodense to isotense, 1 and with contrast there will be patchy pattern of enhancement.2 On the other hand, in T2W1 it is heterogenous. This is due to the presence of tumor cysts, vessels, and calcification. Most tumors enhance in MRI with contrast.1
In Magnetic Resonance Spectroscopy there will be elevated Choline levels, decreased N-Acetyl Aspartate levels 1 2, and increased ration Choline Aspartate. 2
Spinal imaging is done either preoperatively or within 2-3 weeks after the surgery for staging of the tumor. An IV Gadolinium is given with MRI, and a water-soluble contrast with CT or Myelography. These are done to role out drop metastasis.1
CSF analysis, if safe, must be done as well for staging, preferably two weeks postoperatively to prevent any false-positive results.7
*Diagnosis obstacles:
The symptoms of CNS tumors are like those of other common illnesses. Therefore, it is important to demark the clinical picture of tumors to prevent unnecessary investigations and avoid delay in diagnosis.4
Treatment
The management of the discussed tumor lines is “triple therapy”. This includes surgery, radiation, and chemotherapy. Radiotherapy includes a craniospinal axis dose as a result to the high incidence of spinal and leptomeningeal tumor seeding. 2 The treatment protocol begins with surgery, followed by radiotherapy and chemotherapy. 8 The goal of the treatment is to maximize survival and minimize the treatment consequences.7
• Surgery: a standard treatment protocol with the goal to remove as much as possible of the tumor without resulting in neuronal damage. 1 Tumor resection is divided into total resection, subtotal (more than 90% of the tumor is removed), and partial resection (when <90% of the tumor is removed). 4 Depending on the degree of resection, the disease outcome can be determined. 7
When the tumor had attachment in the floor of the 4th ventricle, if there is tumor invasion, or if it is within the area of the Facial Colliculus, it is acceptable to keep residual tumor. In the condition where the posterior arches of C1, and sometimes C2, to reach the Foramen Magnum in midline tumors, the spread of the tumor with the thickening of the arachnoid matter may happen. This is known as “Sugar Coating”.1
• Permanent shunts: they are needed in 30-40% of the patients. 1
• Tumor filters: they are no longer used as frequently as before due to their high incidence of obstruction. 1
• Chemotherapy: when patients are younger than 3 years, chemotherapy can be used to delay radiotherapy in about 20-40% of patients with none disseminated Medulloblastoma.2 Patients that have a tumor relapse will benefit from chemotherapy. 7 However, chemotherapy must be given with radiotherapy even if no indication of disease presence is apparent after the radiotherapy.7
The drugs used include: Cisplatin, Vincristine, Lomustine, Cyclophosphamide, and Etoposide. These drugs are given instantaneously with high dose intrathecal (Methotrexate of Mafosamide), intravenous, or intraventricular Methotrexate. Adults with SHH activated MDB may possibly have an increment in the response to SMO receptor inhibitors, and those drugs that inhibit SHH (Visomodegib).1 Vincristine, Cyclophosphamide, and Cisplatin, and Ectoposide are frequently used in combination. In addition, Intrathecal Methotrexate can be given with Topotecan as part of the treatment plan. Furthermore, Myeloablative Chemotherapy with a hematogenic Stem Cell rescue following it has been used.2 Patients younger than three years, and some who are below 4 years, are given high dose chemotherapy (Vincristine, Cisplatin, Cyclophosphamide) with Thio-peta based higher dose chemo, and peripheral stem cell rescue.3 If sole chemotherapy treatment fails, radiotherapy is started with both CSI and local boost.7
• Radiotherapy: radiotherapy is given to the craniospinal axis, and to the primary tumor site in patients older than 3 years. Patients who are younger than this age will demonstrate delirious sequalae to their developing nervous system. The tumors described above are radiosensitive, and radiotherapy is the best post-surgical therapy. The purpose of the craniospinal axis irradiation (CSI) is to eliminate tumor cells that may remain hidden clinically. A greater dose of radiation is given to the tumor site. Whether primary or a residual tumor in an older child. An added radiation is given to the hypothalamus and the temporal lobes.7
In low-standard risk MDB, particularly those within the pediatric age group, can be treated with a decreased radiotherapy dose of 23.4 Gy to the craniospinal axis. This is in addition to the radiotherapeutic boost of 54-55.8 Gy to the primary site. Adjuvant chemotherapy is used as well. In high risk MDB, conventional fractionated radiotherapy once per day of 54 Gy to the primary tumor, and 18 Gy to the craniospinal axis.1When high-dose radiotherapy is used for a long duration harmful consequence will result with MDBs.2
To decrease the hearing damage, which scatter from the radiotherapy boost to the posterior fossa can cause, intensity modulated radiotherapy is used. Furthermore, a method that needs more studies to explore its efficacy and safety, is the use of proton beam therapy to reduce the toxic effect to none target tissues. 7
The treatment with radiotherapy, with or without chemotherapy, post-surgically is used in patients older than 3 years. 4 This combination of therapy will result in a 5-year survival of 85-95% in a none-disseminated disease, 7 and a progression free survival (PFS) of 81-86%. In younger patients (<3 years), who have MDB, the PFS is 30-40%. Possibly due to the aggressive nature of the disease, and the omission of radiotherapy from the treatment protocol for this age group. However, Desmoplastic MDBs have PFS of 85% at 5 years even when treated with chemotherapy alone. In general, MDBs have better prognosis than the other tumors. 3
The use of biological agents in infants and children is being studied to improve the treatment outcome. 3
• Diet and lifestyle:
A. Evasion of supplemental antioxidants is essential due to the promotion of the reduction in tumor cells death. 7
B. In one third of the cases there will be malnutrition as demonstrated by their calculated BMI. This problem is important to address since malnutrition has multiple disadvantages, among them is the increased rate of side effects of the drugs given for cancer patients. Cyproheptadine Hydrochloride and Megestrol Acetate are helpful in weight gain for patients suffering from cancer and cancer-treatment-related cachexia. However, if weight loss is severe and unrelenting, parenteral, and enteral feeding is necessary. Particularly if there is a neurological deficit that impairs the oral feeding. It is worth mentioning that diet counseling was not proven to be an appropriate management for the malnutrition these patients suffer from. 7
• Physical and speech therapy + exercise: they enhance the outcome in adult patients postoperatively, and they are recommended in children though no studies are present. 3
• In emergency presentation, the patient should be stabilized with placement of extra-ventricular drain and administering high-dose IV steroids. Once the patient is stable, MRI should be done with or without contrast. 7
The best overall survival is in patients who have done total resection, are given radiotherapy, high dose chemotherapy, and autologous stem cell rescue. 3
The combination of Azacytidine with Vorinostat, and Rapamycin have a synergistic impact on ETMR cell line growth. 3
Other promising advances in treatment include the addition of Histone Deacetylase Inhibitor, which is a differentiation agent, with the conventional chemotherapy. In addition, the therapeutics that target the SHH and WNT pathways have been suggested. 3
* Treatment complications:
1) Hair loss: patients receiving chemotherapy and radiotherapy will suffer from temporary hair loss. Depending on the radiotherapeutic dose, the degree of alopecia will occur. Radiotherapy induced alopecia remains until 2-3 months after stopping the therapy. However, full recovery may extend to a couple of months after that. When chemotherapy is taken, hair recovery is after stopping the treatment as well.7
2) Dermatitis that is generated by radiation is treatable with skin moisturizers. Nonetheless, caution should be placed when the patient is taking anti-epileptic drugs (Phenytoin and Carbamazepine) due to their increased risk for developing Steven Johnson Syndrome. The presentation is of a rash that goes beyond the radiation area.7
3) Myelosuppression: it is severer when both radiotherapy and chemotherapy are given in conjugation. Its management is by giving blood, platelets, and/ or GM-CSF for neutropenia.7
4) Mucositis and/or esophagitis could occur due to radiotherapy. they are treated with oral anesthetics rinses (Lidocaine), proton pump inhibitors, and/or H2 blockers.
5) Radiotherapy can result in an increased risk for the development of strokes. In general, the exact incidence of cerebrovascular accident (CVA) is not known, but a patient with brain tumor presenting with clinical symptoms of CVA, or if they have a high risk, should undergo MRA. 7
6) Neurocognitive and endocrine consequences: in one third of school-aged children they will need special education centers. The most common hormone to be affected is growth hormone. Other frequently injured sites within the endocrine system is the hypothalamus, pituitary gland, and the thyroid (hypothyroidism). Hypothyroidism results from scatter from CSI. 7
7) 2 to 8 weeks after radiotherapy, patients may suffer from fatigue, increased sleepiness that reaches up to 18 hours, nausea, and anorexia. This is known as Somnolence Syndrome. On investigations, there will be electroencephalography slowing and rarely CSF pleocytosis. Only reassurance is required. 7
8) Moya-Moya disease: this could occur years after radiotherapy.7
9) Cisplatin that is given with radiotherapy can result in hearing loss. 7
10) Surgery complications: septic or aseptic meningitis, CSF leak, pseudomeningiocele, diplopia, Abducens nerve injury in Bone Marrow resection (temporary for a period of a couple of months), ataxia in resection of the Vermis, incoordination of the limbs in Cerebellar Peduncle injury. 7
*Prognosis:
Poor prognostic factors include those that are related to age (1.5 cm postsurgical residual tumor). 1 Other poor prognostic factors include postponement in diagnosis, insufficient initial response to treatment, a general condition that is poor, great tumor size, and poor defined margins. The median survival is less than 2 years in ATRT. 2 On the other hand, the five-year survival for MDBs is up to 70%. 8
Appropriate diagnosis with accurate molecular identification results in a better outcome. 2 When diagnosis is delayed, there will be tumor evolution, development of hydrocephalus, and tentorial herniation. If the delay reached to the point of requiring prompt surgical resection, delirious sequelae to the neurocognitive and endocrine function, surgical morbidity, and visual outcome. 4
If the patient has disseminated disease and an incomplete surgical resection is done, the outcome will be poor. 3 MDB usually has wide invasion, which requires limitation to the resection of the tumor (incomplete resection). In MDBs, drop metastasis should always be assessed for. In addition, MDBs have seeded the craniospinal axis in 10-35% of the cases, and in 5% extra-neural metastasis. This is sometimes promoted by shunting, even though shunts are uncommon. Children and infants are the most difficult to manage due to the aggressive nature of the disease, and their young age. 1
CSI has shown better prognosis, while giving chemotherapy prior to radiotherapy in patients older than 3 years has worse outcome. 3
When secondary tumors happen (Meningioma and Glioma) they have worse mortality and morbidity. 7
* Tumor type specific treatment and prognosis:
1) Medulloblastoma: due to the varying liquid biopsy biomarkers, treatment is impaired in MDBs. After standard treatment, several patients with MDBs result in recurrence of the tumor, or distant metastasis. 40% will have metastasis at the time of diagnosis, and in more aggressive tumors they can have local recurrence or metastasis after treatment.
MDBs are very heterogenous on both cellular and molecular basis. However, the presence of stem cells that are capable of self-renewal is an indicator for possible post treatment recurrence. These cells will either be normal stem cells that have undergone malignant changes, or they can be neoplastic cells that have acquired stem-cell properties by their collaboration with the extracellular factors, and microvesicles with the microenvironment surrounding the tumor.
Important prognostic factor for MDBs is the expression of c-erbB-2 (HER2/neu) oncogene. (radiopedia/ medulloblastoma)
The most common MDB subtypes that are associated with poor prognosis due to the recurrence after treatment (surgical, radiotherapeutic, and chemotherapeutic) are SHH subtype, group 3, and group 4. Both SHH and group 3 are associated with 70% recurrence in a year. The recurrence in groups 3 and 4 is the hardest to treat. 6
2) ETMR: it has a uniform dismal prognosis even with ultimate multidisciplinary management.9 10 Optimal treatment protocol is not yet identified.9,10 However, the combination of surgical resection, CSI, and chemotherapy is advised by several doctors. To enhance the outcome, it has been suggested that extensive resection of up to 1 cm should be done. This is performed on the tumor’s nearby brain tissue, or infiltrated tissue. Extended surgical resection method includes the use of high dose chemotherapy and decreased prophylactic CSI dose. Using this management protocol was reported to encompass enhanced outcome as compared to giving chemotherapy and high dose chemotherapy alone post operatively. Nonetheless, since most ETMR patients are younger than 4 years, the impact of CSI on the neurocognitive function must be kept in mind. 9
At diagnosis, some cases had leptomeningeal invasion, invasive involvement of the extracranial soft tissue, and metastasis. If not present at the time of diagnosis, these features were developed in several cases with time. A recent study showed that tumors that recur after treatment show increment in somatic single nucleotide variant, which were discovered to be associated with a mutation that occurs as a result to the exposure to Platinum based agents. This implies possible association with treatment; a finding that can explain the reason behind rising chemotherapeutic resistance and suggests the importance of early radiation.
Studies show that the percentage of metastasis at diagnosis is 13%, encouraging the possibility of using radiotherapy that is focused on tumor site only, if given early. In addition, the poor sequalae after radiation can be decreased to a limited degree based on the use of focal, low radiation exposure, and proton therapy.
Intrathecal administration of either Etoposide alone, or Etoposide used interchangeably with liposomal or aqueous Cytarabine with/without Topotecan was shown to decrease the leptomeningeal spread in patients receiving radiotherapy alone. Inappropriate for this management approach are infants aged younger than 18 months, and infants with metastasized tumor initially or those with significant are of treatment or volume. Treatment option for this group is the use of early intensive chemotherapy (maintenance therapy following a high dose chemotherapy) using new agents.
Generally, they are treated with surgery followed by chemotherapy with or without delayed radiotherapy.10
3) AT/RT: in adults, it has high recurrence rate after surgery. Their prognosis is poor, even after the triple approach of surgery+ radiotherapy+ chemotherapy. Nonetheless, the triple approach is better than other treatment approaches in terms of outcome. 16
In younger children, giving intensified chemotherapy as an alternate to radiotherapy to avoid the delirious toxic effect of radiotherapy in this age group.17 The treatment for AT/RT has no specific approved tumor management protocol. 17