Article topic: Dandy Walker Syndrome
Author: Ghayda Netham Al-Majali
Scientific editor: Dr. Omar Jbara
Linguistic editor: Philip Sweidan
Overview
Dandy-Walker syndrome (DWS), or Dandy-Walker malformation (DWM), is a rare congenital brain malformation that occurs during embryonic development of the cerebellum and fourth ventricle. It was first described by Dandy & Blackfan in 1914 and dubbed “Dandy Walker malformation” forty years later by Benda (1). DWS is the most common posterior fossa abnormality characterized by agenesis or hypoplasia of the cerebellar vermis and cystic dilation of the fourth ventricle causing enlargement of the posterior fossa with cerebrospinal fluid and upward displacement of the tentorium and torcula (2)(3). Patients may have hydrocephalus at the time of diagnosis (4).
Embryology
The rhombencephalon (hindbrain) develops into the metencephalon (pons and cerebellum) and myelencephalon (medulla oblongata) around the fifth week of gestation. Development of cerebellum begins in the ninth week, when the cerebellar hemispheres develop from the rhombic lips, subsequently fusing to form the vermis. The choroid plexus of the fourth ventricle as well as the foramina of Luschka and Magendie form around the tenth week of gestation from the rhombic vesicle. Cerebrospinal fluid (CSF) then accumulates within the fourth ventricle, forming this space. Subsequently, the cerebellar lobules develop in an anterior-to-posterior direction and are completely formed by week 18. Because it develops more slowly than the cerebral hemispheres, the cerebellum appears smaller at 20 weeks gestation relative to the large posterior fossa CSF spaces, leading to a potential over-diagnosis of vermian hypoplasia by antenatal ultrasonography (5).
Etiology and Pathogenesis
DWS was believed to be caused by abnormality and atresia of the foramina of Luschka and Magendie, leading to enlargement of the fourth ventricle and vermian hypoplasia.
However, recent evidence indicates that DWS results from developmental abnormalities affecting the roof of the rhombencephalon, leading to variable degrees of vermian hypoplasia and cystic enlargement (6).
Although the majority of cases are rare and not familial (6), some may result from chromosomal aberrations, Mendelian disorders, environmental exposures (such as German measles and congenital rubella which can severely affect early embryonic development), teratogens, or fetal exposure to alcohol (7).
Although DWS is typically sporadic, first-degree relatives have a higher risk of acquiring DWM compared with the general population (7)(8). Rare genetic mutations in a small number of cases have been detected in some genes including FOXC1, FGF17, LAMC1, NID1, ZIC1, and ZIC4 (8).
There are two hypotheses for the etiology of DWM. The first is the arrest of the vermian development and the second is failure of fourth ventricle foramina fenestration resulting in an enlarged Blake’s pouch, compressing the vermis(9).
Clinical Presentation and Complications
Many patients of DWS remain clinically asymptomatic for years, while others present with numerous comorbidities & anomalies which increase the chances of earlier diagnosis (7).
DWS may be isolated or associated with facial malformations (e.g. angiomas, cleft palate, macroglossia, and facial dysmorphia), musculoskeletal malformations, cardiovascular anomalies (e.g. septal defects, patent ductus arteriosus, aortic coarctation, dextrocardia), ocular abnormalities (e.g. coloboma, retinal dysgenesis, microphthalmia) and genitourinary, respiratory or gastrointestinal system abnormalities (6). Central nervous system (CNS) disorders related to DWS include malformations of cortical development, holoprosencephaly, agenesis of the corpus callosum (ACC) and neural tube defects(2). DWS is also associated with other systemic anomalies including genetic aberrations, environmental factors, teratogens, Mendelian disorders, syndromic malformations, congenital infections, and various other comorbidities (9).
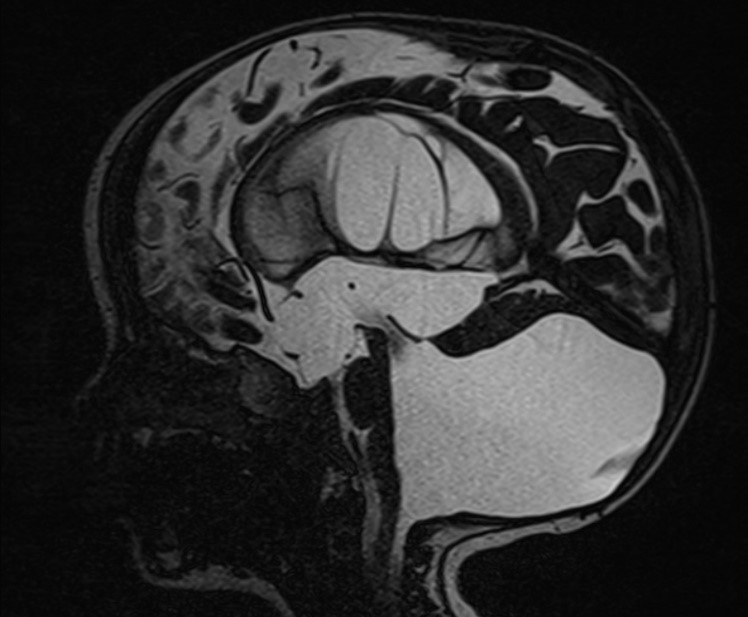
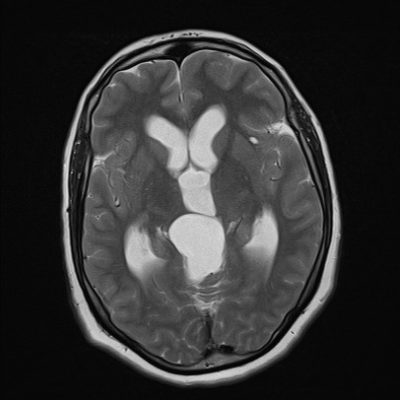
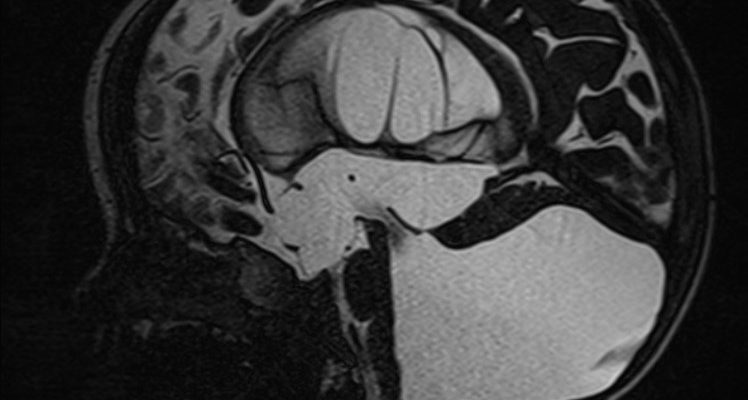
Figure 1: Dandy Walker Malformation ; Case courtesy of Dr Mostafa El-Feky (10).
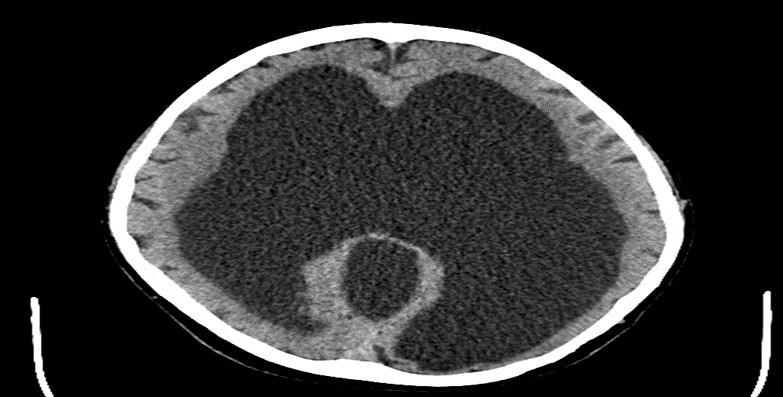
Figure 2: Dandy Walker Syndrome; Partial hypoplasia of the cerebellar hemispheres. Marked expansion and dilation of the ventricles of the brain, reducing the volume of the substance of the brain, hypoplasia of the corpus callosum; Case courtesy of Dr Osadchiy Anton (11).
Differential Diagnosis
Differential diagnoses include posterior fossa abnormalities including retrocerebellar arachnoid cysts, cystic hygroma, Blake’s pouch cyst (BPC), mega cisterna magna, and vermian hypoplasia. These may be sometimes associated with hydrocephalus, which can commonly present in a similar way to DWS (6).
Additionally, several syndromes are correlated with DWS, such as Aase-Smith, cerebro-oculo-muscular syndrome, Coffin-Siris, Cornelia de Lange’s syndrome and Aicardi, Fryns syndrome, Klippel-Feil syndrome, Turner’s syndrome and Joubert’s syndrome which is absence or underdevelopment of the cerebellar vermis (2)(7).
Retrocerebellar arachnoid cysts might be large enough to cause compression of cerebellar hemispheres and fourth ventricle which can produce significant mass effect and the vermis remains intact (2).
BPC presents as a retrocerebellar fluid collection characterized by posterior ballooning of the superior medullary velum into the cisterna magna, below and posterior to the vermis with communication to the fourth ventricle (2).
The typical finding in mega cisterna magna is an enlarged cisterna magna measuring more than 10mm in the axial plane at the level of the transcerebellar diameter. However, fluid collection is located postero-inferior to a normally developed cerebellum with no mass effect on it, normal vermis and normal fourth ventricle (2).
Dandy-Walker variant (DWV) is a term first introduced by Harwood-Nash and Fitz to describe a milder spectrum of DWS-like signs that did not correspond with the classic definition. DWV consists of an inferior cerebellar vermian defect and communication between a normal-sized cisterna magna and fourth ventricle so it does not fit all of Dandy Walker Syndrome criteria for diagnosis (e.g. vermian hypoplasia and cystic dilatation or enlargement of the fourthth ventricle, without enlargement of the posterior fossa)(5).
Dandy Walker Complex (DWC) is another expression describing a continuum of posterior fossa malformations categorized from mild (mega cisterna magna only) to moderate (hypoplasia of vermis, enlarged fourth ventricle) to severe (agenesis of vermis, dilation of posterior fossa cyst and fourth ventricle) (12).
So, DWS requires six radiographic features for diagnosis:
- Large posterior fossa cyst communicating with the 4th ventricle.
- Absence of a portion of the inferior vermis at different degrees.
- Hypoplasia, anterior rotation, and upward displacement of the vermis remnant
- Absence or flattening of the angle of the fastigium (highest point of fourth ventricle).
- Large posterior fossa with torcular elevation.
- Anterolateral displacement of normal or hypoplastic cerebellar hemispheres (5).
This wide spectrum of posterior fossa cystic anomalies provides a basis for differential diagnosis in prenatal or postnatal presentations and these contain Dandy Walker Variant , mega cisterna magna, posterior fossa arachnoid cyst, persistent Blake’s pouch cyst, fourth ventriculocele, and congenital vermian hypoplasia(5).
Differentiating features
DWS and DWV are difficult to distinguish, and may act as a continuum of developmental abnormalities grouped together as Dandy Walker complex (DWC)(1). Retrocerebellar arachnoid cysts and BPC may imitate and mimic DWS, but do not have vermian agenesis (1). The position of the choroid plexus of the fourth ventricle is normal in arachnoid cysts, absent in DWS, and displaced into the superior cyst wall in BPC (1). An intrathecal enhanced Computed Tomography (CT) scan (which performed after instilling iodinated contrast into a ventricular catheter) would distinguish and identify a mega cisterna magna which communicates with the ventricles, while DWS and most (but not all) arachnoid cysts do not (1).
 Figure 3: Dandy Walker Variant (DWV); Case courtesy of Dr Aditya Shetty {A cystic area of CSF density is seen in the posterior fossa, with a “keyhole” appearance. This cystic area is in continuation with the 4th ventricle. Vermian hypoplasia is also seen with gross hydrocephalus. The “keyhole connection sign” (white dashes) indicate to the abnormal connection of the cistern Magna and the posterior aspect of the 4th ventricle} (13).
Figure 3: Dandy Walker Variant (DWV); Case courtesy of Dr Aditya Shetty {A cystic area of CSF density is seen in the posterior fossa, with a “keyhole” appearance. This cystic area is in continuation with the 4th ventricle. Vermian hypoplasia is also seen with gross hydrocephalus. The “keyhole connection sign” (white dashes) indicate to the abnormal connection of the cistern Magna and the posterior aspect of the 4th ventricle} (13).

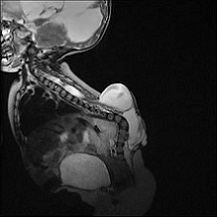
Figure 4: Blake’s Pouch Cyst (BPC) – Axial T2; Case courtesy of Dr Sepehr Haghighi; {5-year-old female presented with Headache. The enlarged 4th ventricle is seen freely communicating with infracerebellar posterior fossa cyst. The vermis of cerebellum is upward displaced with no hypoplasia. Normal bidirectional flow of CSF at the cranio-cervical junction and cervical subarachnoid spaces is detected} (14).

Figure 5: Mega Cisterna Magna – Axial T2; Case courtesy of Assoc Prof Frank Gaillard (15).
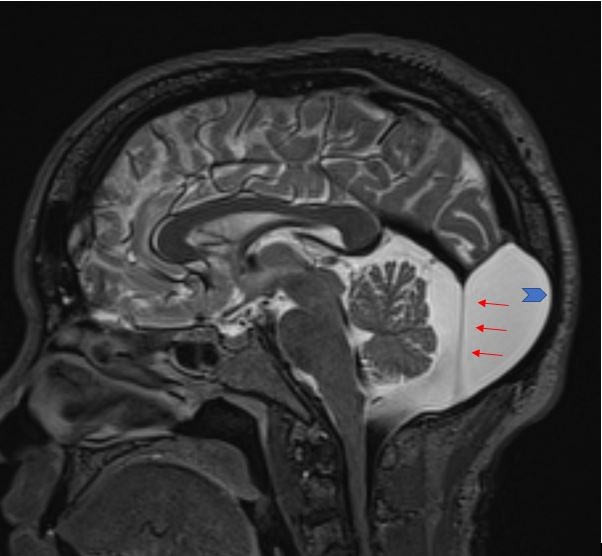
Figure 6: Retro-cerebellar Arachnoid Cyst- sagittal T2; Case courtesy of Dr Amr Farouk. {Annotated image showing the membrane of the arachnoid cyst (red arrows). Remodeling of the skull is also present (blue arrow)}(16).
Epidemiology
DWS and related variants have an incidence of 1 in 35000 live births in the United States (1). It is reported in only 1 in 25000 to 30000 live births which occurs during the embryonic development of the cerebellum and 4th ventricle in Asia (7).
Autosomal recessive inheritance has been determined in a few cases, Male: female ratio is 1:3 and it may be associated with PHACES syndrome, which stands for Posterior fossa malformations, hemangioma, arterial lesions, cardiac abnormalities, and eye abnormalities (1). These anomalies account for approximately 1 to 4% of Hydrocephalus cases (2). This malformation is found in 4% to 12% of hydrocephalus in infants (17).
Treatment of hydrocephalus with shunting improves mortality (18). Fetal mortality directly correlates with the presence of extra CNS malformations (19). Gestational exposure to rubella, CMV, toxoplasmosis, Warfarin, alcohol, and isotretinoin are thought to be risk factors(1).
Prognosis
Patients with DWS may present different degrees of hydrocephalus (approximately, 80% of DWS cases have hydrocephalus), which if untreated may lead to severe neurological abnormalities and death (6). 50% of Children with untreated hydrocephalus die before the age of 3 years, and only around 20 to 23% reach adult life or puberty (6). Most patients with untreated hydrocephalus who reach adult life will have motor, visual and auditory deficits (20). The diameter of the fetal lateral ventricle through obstetric ultrasound may have important prognostic value, and as described in a previous study, lateral ventricles measuring between 11-15 mm are associated with a 21% risk of delayed development (6). If the diameter measures more than 15 mm, the risk of delayed development is above 50% (21). Most case series of patients with non-tumoral hydrocephalus may indicate a risk for epilepsy of approximately 30% (20).
Functional outcomes vary due to several factors, which include other structural brain anomalies, extra-CNS manifestations, epilepsy, motor, visual or hearing impairment, and other congenital anomalies (22)(23). Therefore, the prognosis differs widely as there are different levels of severity of the malformation (1). Pediatric neurosurgical literature suggests a 12–50% of mortality rate (24), although this is improving with modern shunting techniques (1). Ataxia, spasticity or alteration in muscle tone, and poor motor control are also common and only 50% have normal IQ (1).
In (10%-20%) of cases with DWS, manifestations, signs and symptoms of the malformation do not appear until late childhood or adulthood. These patients have a different range of features compared to those affected in infancy. These features could include headaches, an unsteady walking gait, facial palsy, increased muscle tone, muscle spasms, and mental and behavioral changes. Very rare that people with DWS have no health problems related to the malformation. Problems related to hydrocephalus or complications of its treatment are the most common cause of death in people with DWS (25).
Treatment and Management
Treatment of DWS is through treating the manifestations and associated comorbidities (6). Most patients present with signs and symptoms from increased intracranial pressure (ICP), most commonly related to hydrocephalus and posterior fossa cyst. For this reason, treatment generally aims to control intracranial pressure, usually through surgery with a focus on alleviating hydrocephalus and posterior fossa symptoms (2)(4). Treatment may include surgical interventions like ventriculoperitoneal (VP) shunting, or cystoperitoneal (CP) shunting. Other patients may be candidates for endoscopic procedures, including endoscopic third ventriculostomy (ETV) (6).
Ventriculoperitoneal (VP) shunt
A VP shunt is a cerebral shunt that drains excess and accumulated CSF when there is an occlusion in the normal outflow, or decreased absorption of the fluid (26). Accumulated CSF can increase the intracranial pressure resulting in cerebral edema and ultimately herniation (26). The shunts drain excess accumulated CSF into the peritoneal cavity, the atrium, or the pleura; thus, appropriately called ventriculoperitoneal, ventriculoatrial, and ventriculopleural shunts (26).
Indication of VP shunt
Disorders typically requiring shunting include:
- Congenital hydrocephalus after aqueductal stenosis,
- Tumors leading to CSF blockage of the lateral ventricles, third ventricle, and those in the posterior fossa blocking the cerebral aqueduct or fourth ventricle,
- Communicating hydrocephalus secondary to meningitis or subarachnoid hemorrhage,
- Myelomeningocele because the flow of CSF is altered due to hindbrain malformation,
- Idiopathic intracranial hypertension,
- Arachnoid cysts,
- Dandy-Walker syndrome with a cystic deformity of the fourth ventricle, hypoplasia of the cerebellar vermis, and an enlarged posterior fossa (26).
Contraindication of VP shunt
Ventriculoperitoneal shunt is absolutely contraindicated with infection over the entry site, infection of the CSF and allergy to any of the catheter components (27). It is relatively contraindicated in these cases: altered coagulation function, high CSF protein and CSF with blood (26).
Equipment Needed for Surgery
A VP shunt is placed in the operating room under general anesthesia by a neurosurgeon. The equipment needed to perform the procedure include:
- Shunt system (ventricular catheter, peritoneal catheter, and small, medium or high pressure valve), specimen tubes for CSF,
- Drill for the burr hole,
- Shunt passer
- Image-guided navigation system only for small ventricles (26).
Preoperative Preparation
Brain magnetic resonance imaging (MRI) or computed tomography (CT) scan is reviewed for the planning of the shunt and proper placement of the proximal catheter. The patient is placed supine, for a parieto-occipital approach, the head is tilted to the contralateral side of the shunt placement. Preoperative antibiotics are given, and the patient is dressed in sterile conditions (26).
The Technique of VP shunt
The Technique of VP shunt is done by shaving the hair away from the head incision site, cleaning the skin with an antiseptic and applying a sterile fenestrated drape over the incision sites (head, neck, chest, and abdomen) (26). Then, making a “U or C” shaped skin incision over the entry point where the burr hole is to be performed for the introduction of the ventricular catheter (28).
If the frontal approach will be used, Kocher’s point is used; which is an entry point that is 11 cm superior and posterior from the nasion, 3 cm lateral to midline along the mid pupillary line, and 1-2 cm anterior to the coronal suture; catheter should then be passed to a depth of 5-5.5 cm (28).
If parieto-occipital approach will be used, Keen’s point is used; which is approximately 2.5 to 3 cm superior and posterior to the pinna and the catheter should be passed to a depth of 4 to 5 cm or until reaching the trigone of the ipsilateral or the same side lateral ventricle, but sometimes it is aiming toward the frontal horn of the lateral ventricle so Alternatively, Dandy’s point can be used which is 3 cm above the inion and 2 cm left or right to the midline (28).
Burr hole is done at the desired entry point and the dura incised, a small entry point in the cortex is coagulated and incised, ventricular catheter is introduced directed into the ventricle and cut to the appropriate pre-measured length, then CSF samples are collected and the ventricular shunt is connected to the valve and secured with a silk tie (26).
An incision in the abdomen is done to access the peritoneal cavity; the site depends on the surgeon’s preference and can be done in the upper quadrant or the midline and Shunt passer is used to pass the peritoneal distal catheter between both incisions (26).
Peritoneal catheter is connected and attached to the valve and secured with a silk tie and after good distal CSF flow at the peritoneal catheter, it is introduced into the peritoneal cavity. Finally, Wounds are closed in anatomical layers (CSF sample is sent to the laboratory for cell count, glucose, protein, gram stain, and culture) (26).
Complication of VP Shunting
Complications include:
- Infection due to skin flora entering the shunt (usually from Staphylococcus epidermidis),
- Intracerebral or intraventricular hemorrhage,
- Malposition,
- Shunt disconnection or obstruction,
- Subdural hematomas. (26).
- Abdominal CSF collections (pseudocyst or CSFoma)(29).
Acute signs and symptoms of malfunction or infection include: headache, lethargy, diplopia, nausea, vomiting, seizure, irritability, poor feeding, head enlargement when sutures are open, fever and neck rigidity (26).
Clinical Significance, Evaluation and Outcome
Tapping a shunt is performed for both diagnostic and therapeutic reasons, and VP shunt is considered one of the advances that has a high impact on neurosurgical patient care (26).
Shunt X-rays are done to evaluate the integrity of the system and CT scan or MRI is done to evaluate the size of the ventricles (26). VP shunt can be lifesaving, but the eventual outcome depends on the reason why the shunt was inserted (26).For benign or mild disorders, most patients have a good outcome, but for malignant tumors the outcome is usually poor; often patients die from other causes unrelated to the shunt (26).The complication rates of VP shunts range from 2-20%. In addition, shunt revision is required in about 5-10% of neonates and pediatrics (30).
Updated Researches Regarding Management/ Treatment
Treatment options for Dandy-Walker malformation – Retrospective Study – Published in Nov/2006 in the journal of neurosurgery (JNS)
DOI: 10.3171/ped.2006.105.5.348
The study aimed to evaluate the effectiveness of various treatment options available for children with DWM and the role of endoscopic procedures (ETV) in the treatment of this syndrome (31) by retrospective review of 72 children who underwent surgical treatment for DWS during a sixteen-year period. All patients underwent CT scanning, and 26 underwent MRI. The initial surgical treatment included (VP) shunt insertion in 21 patients, (CP) shunt insertion in 24 patients, and (combined VP and CP shunt) insertion in 3 patients, and 21 patients underwent endoscopic interventions (ETV alone in 16 patients, ETV with aqueductal stent placement in 3 patients, and ETV with fenestration of the obstructing membrane in 2 patients). Also, 3 patients underwent membrane excision via a posterior fossa craniectomy (31).
In the 26 patients who had undergone preoperative MRI, aqueductal patency was noted in 23 patients versus aqueductal obstruction in 3 patients. These three patients underwent insertion of a stent from the third ventricle to the posterior fossa cyst in addition to the ETV surgical procedure. During the follow-up period, 12 patients with a CP shunt and 4 patients with a VP shunt experienced shunt malfunctions and complications that required revision. Four patients with a CP shunt also required placement or insertion of a VP shunt. In addition, five of the 21 ETVs failed, requiring VP shunt placement (31).
A reduction in the size of ventricle was noted on postoperative images occurred more frequently in patients with VP shunt, whereas a decrease in cyst size was more obvious in patients with CP shunt. Successful ETV resulted in a slight decrease in ventricle size and varying degrees or levels of reduction in cyst size (31). Therefore, it was concluded that ETV may be considered an acceptable alternative in children with DWS (31).
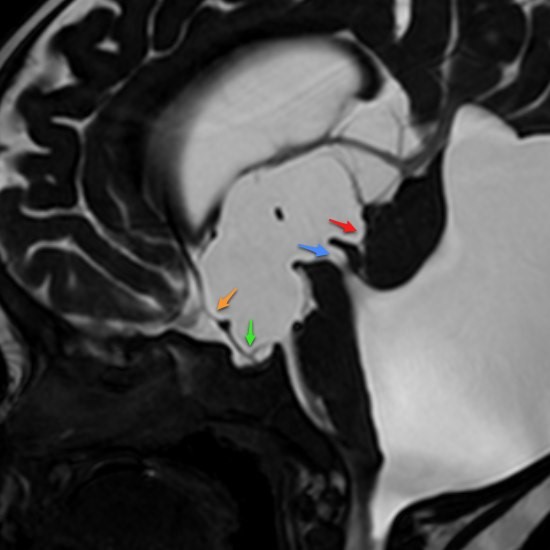
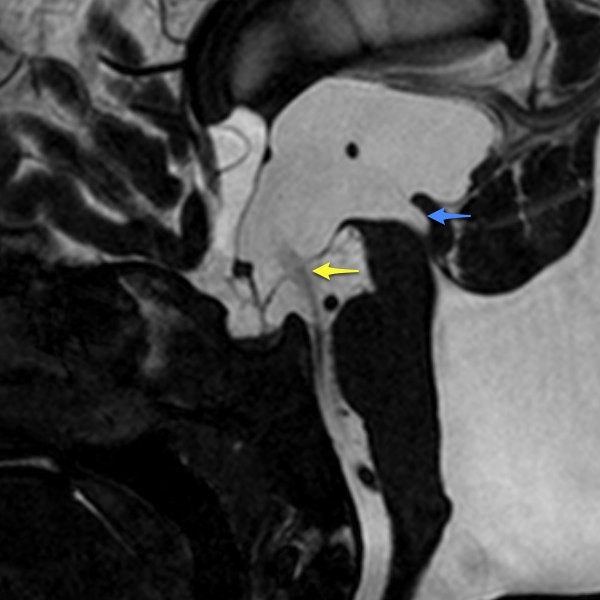 Figure 7: (A); pre ventriculostomy, (B); post ventriculostomy. { Endoscopy Third Ventriculostomy; Case courtesy of Assoc Prof Frank Gaillard} (32).
Figure 7: (A); pre ventriculostomy, (B); post ventriculostomy. { Endoscopy Third Ventriculostomy; Case courtesy of Assoc Prof Frank Gaillard} (32).
Finally, for further knowledge about surgical interventions, here are some links:
Ventriculo peritoneal shunting: (33)
- https://www.neurosurgicalatlas.com/volumes/csf-diversion-procedures/ventriculo-peritoneal-shunt
- https://www.youtube.com/watch?v=ZSV1LauJlYk
- https://www.youtube.com/watch?v=vbwonC23oro
- https://www.youtube.com/watch?v=SDsBr6sXkH4
Endoscopic Third Ventriculostomy: (34)
- https://www.neurosurgicalatlas.com/volumes/csf-diversion-procedures/endoscopic-third-ventriculostomy
- https://www.youtube.com/watch?v=18J1hpG7G7U