Article Topic: Tuberous Sclerosis.
Author: Shahd Etoom.
Editor: Rahmeh Adel.
Reviewer: Ethar Hazaimeh.
Keywords: Tuberous sclerosis complex (TSC); Dermatology; Sirolimus; Lymphangioleiomyomatosis (LAM); RAS homolog enriched in brain (Rheb); Mammalian target of rapamycin (mTOR).
Abstract
Tuberous Sclerosis Complex (TSC), sometimes called Epiloia or Bourneville-Pringle disease, is an autosomal dominant neurocutaneous syndrome with a range of clinical manifestations. It is a multisystem condition that could be randomly linked to hamartomas in several organs. Dermatologists greatly aid the history of the condition, as the most common finding is cutaneous involvement, which allows for early diagnosis of the syndrome and intervention throughout its natural course. The history of this ailment has been altered by recent discoveries on the encouragement of tumor growth, opening the door to topical and systemic pharmacological therapy studies.
Since tumor replacement can cause severe morbidity and mortality, understanding these themes can help manage impacted patients more effectively.
Introduction
Tuberous Sclerosis, often called Bourneville Pringle disease or Epiloia, is an autosomal dominant neurocutaneous syndrome exhibiting a range of clinical manifestations. It’s a multisystem illness that could be randomly linked to hamartomas in different organs. Skin and the central nervous system are the most commonly affected. [1]
Isolated case reports from centuries ago detailed some of the different clinical signs brought on by a pathology that was unknown at the time. Virchow and Von Recklinghausen discovered hamartomas in the heart and brain, respectively, in the middle of the 1800s. [2] The word “tuberous” was first used in a post-mortem analysis of the brains of individuals who had convulsions and cognitive impairment because the tumors resembled tubercular foods like potatoes.
The results were simultaneously published by Bourneville, who was the first to report the discovery of a new syndrome, only in the late 1800s. [3]
Even while other writers had previously reported cutaneous signs, it was not until the early 20th century that they began to be connected to the illness that Bourneville had described. The association between mental retardation, epilepsy, and sebaceous adenoma—the trinity that characterizes TSC—was initially identified by Vogt three years after it was discovered by Campbell in 1905. [4]
Following years of research focused on a few clinical and genetic aspects of TSC, the field of molecular biology came of age. The signaling pathways of hamartoma development are the new objects of investigation. Recently, there has been significant progress in identifying the proteins, enzymes, and signals that are involved in the etiopathological process of the syndrome.
These findings are applied to the understanding of TSC, as it shares unchecked tumor cell development as a common cause. Due to the significant progress made in this field, clinical trials of novel medications that have the potential to modify the path of tuberculosis—a disease that causes significant morbidity and mortality—can now be conducted, two centuries after Virchow and Von Recklinghausen first described tubers. [5, 6]
Epidemiology
Diverse conclusions are drawn from studies evaluating the prevalence and incidence of tuberous sclerosis complex. Due to its high phenotypic heterogeneity, the condition can be difficult to diagnose. [7] One in 10,000 babies is affected, and most patients receive a diagnosis within the first 15 months of their lives. [6, 8]
Of those diagnosed beyond that age, about 25% had previously displayed distinctive clinical indications that were disregarded in an earlier medical evaluation. [4, 8] We now have an estimated frequency of 1:6,000 people in the general population due to the development of new genetic research techniques and the identification of novel clinical symptoms. [6]
Although the incidence of symptoms is similar for both sexes, women may exhibit more overt symptoms. Reports demonstrating disproportionate participation in a specific ethnic group are nonexistent.[9]
The presence of early-onset seizures is the symptom that most frequently guides a diagnosis of the condition. [6, 8] The diagnosis is also aided by the involvement of the skin and mucous membranes; nevertheless, the characteristic skin signs typically occur later in life. While there is variation in the occurrence of illnesses related to certain organ systems, the primary causes of death and morbidity are related to neurological and renal problems.[10]
Pathogenesis
The autosomal dominant condition known as tuberous sclerosis complex has a high penetrance. TSC1 and TSC2 tumor suppressor genes, which are found on chromosomes 9q34 and 16p13.3, respectively, are the cause of it through inactivating mutations. [11] The TSC1 gene oversees encoding the protein hamartin, while the TSC2 gene encodes tuberin. The hamartin-tuberin combination is a significant tumor growth inhibitor. Loss of inhibition on cell migration and proliferation is brought on by its absence. [3] Many genetic alterations are seen, essentially affecting every exon of the relevant genes, including nonsense, missense, and deletion mutations. [6] Fifteen percent of cases have no mutations found, despite the changes being verified.[3]
Although hereditary transmission may result in the familial types of TSC, somatic mutations account for 70% of cases, particularly when TSC2 is the mutated gene. These alterations are caused by germline mutations. Sporadic incidences of the disease are caused by somatic mutations. [6] The wide range of mutations and potential gene involvement contribute to the partially explained phenotypic variability of the illness. The occurrence of mosaicism is another aspect that could account for the unexpected nature of TSC clinical presentations. [12, 13]
Research indicates that in occasional occurrences, mutations in the TSC2 gene are observed to happen five times more often than mutations in the TSC1 gene in sporadic cases. Whereas in familial transmission these changes occur with equal frequency (1:1 ratio). [6]
Compared to TSC originating from mutations in TSC1, changes in the TSC2 gene cause more severe disease. [14-16] Familial transmission cases, in which TSC1 gene mutations occur more frequently, cause mild to severe disease and occasionally fail to meet diagnostic criteria.[16] The “two-hit” theory, put forth by Knudson in the 1970s, provided an explanation for the etiopathogenesis of hamartoma formation in TSC for several years. The theory states that during germ-cell division, persons with TSC would already have a malfunctioning tumor suppressor gene in one of the alleles of TSC1 or TSC2.
The “first hit” would come at this point. However, as both alleles must be deleted or deficient for clinical manifestation to occur, the prior alteration would not be sufficient to cause phenotypic results. Thus, for a hamartoma to occur in TSC, a second alteration involving the normal allele that results in loss of heterozygosity is required. This would be the “second hit”, which occurs during somatic cell growth. According to Knudson, the anomalous allele causes loss of heterozygosity and interferes with the tumor suppressor gene’s ability to function by causing genomic instability or inducing epigenetic events (genetic alterations brought on by non-mutational sources, such as DNA methylation). [17] The absence of the hamartin-tuberin complex as a result of this genetic alteration would cause unchecked cell proliferation. [18, 19]
Recent research has cast doubt on this theory as it was discovered that cancers could occur even in the presence of an intact normal allele. [20] It is unclear exactly how these mutations cause cellular hyperproliferation and the formation of multiple hamartomas anywhere in the human body. This proliferation pathway has already had some of its steps identified. The human body’s cells are constantly responding to a vast array of interconnected biological stimuli. Proliferative diseases including cancer and TSC are caused by malfunctions in this signaling system. Via the phosphoinositide 3-kinase signaling pathway, the TSC1 and TSC2 genes play a significant role in controlling cell growth by blocking the mammalian target of rapamycin (mTOR). As a key sensor of energy and nutrition availability, mTOR controls the proliferation, differentiation, and growth of cells. [5, 21]
As was previously established, the 200Kda protein tuberin is encoded by the TSC2 gene, and the 130Kda protein hamartin is encoded by the TSC1 gene.[22] The signaling protein referred to as “RAS homolog enriched in brain” (Rheb) is inhibited by the hamartin-tuberin complex, which forms a heterodimer.[6] mTOR, in turn, is stimulated by Rheb and controls the phosphorylation of the translation-inhibiting protein 4E-BP1 (PHAS-1), as well as the isoforms of ribosomal protein S6 kinase 1 and 2 (S6K1 and S6K2).[21, 23-25]
The loss of the hamartin-tuberin complex’s inhibitory function on Rheb enables mTOR to stimulate the S6K and 4E-BP1 proteins, increasing ribosomal biogenesis and protein production. This ultimately results in unchecked tissue growth and cell proliferation, which is why tumors are seen in tuberous sclerosis.[26-29] Although its function in controlling biological processes is still unclear, TSC1 and TSC2’s characteristics are of great interest. One such result is that the cyclin-dependent kinase inhibitor p27 is regulated by the hamartin tuberin complex, which plays a significant role in the stages of cell differentiation. This information relates to the biology of the cell cycle.[30]
A further crucial role for loss of inhibition of the mTOR signaling pathway is in the insulin-dependent cellular metabolism. In addition to increasing the risk of apoptosis, mTOR activation causes endoplasmic reticulum stress, which leads to severe insulin and IGF1 resistance.[5] Genetic modifications distinct from those affecting TSC1 and TSC2 can account for other clinical symptoms. The most notable instance is the co-occurrence of polycystic kidney disease and TSC, which is a significant contributor to end-stage renal disease in these individuals. Given that the two mutant genes causing the two diseases are close to one other, they could be regarded as contiguous gene syndromes.
Two sizable deletions in the TSC2 gene cause the PKD1 gene, which causes polycystic kidney disease, to mutate as a consequence, which accounts for the simultaneous presence of both disorders. The locations of both genes are on chromosome 16. [31-34]
Clinical features
Clinical manifestations of tuberous sclerosis arise from the development of hamartomas in different organs. The development of diverse forms of phenotypic TSC is attributed to the dependence of these lesions on the distinct genetic alterations present in the pathophysiology of the disease.[4]
Skin manifestations
Skin clinical manifestations are the most common findings in TSC, despite the fact that some patients do not exhibit skin involvement, according to a study that looks at clinical indicators on the skin depending on age group. [35]
The most common clinical indication of these skin lesions is the existence of hypomelanotic macules (Figure 1). 90 to 98% of individuals have hypochromic or achromic macules, which are particularly common before puberty and sometimes the only symptom in young patients. [3, 35] Such macules, which typically spare the face and mostly affect the trunk and limbs, are reported by parents as existing at birth or in the early stages of life. Throughout life, these macules tend to get larger and more numerous. As adults, they become more pigmented and may even vanish, making them less noticeable.
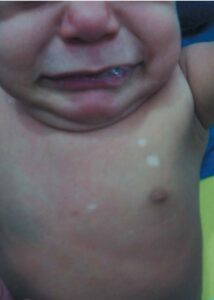
FIGURE 1: Early Findings hypomelanotic macules
Poliosis may occur when achromic macules afflict the scalp.[35, 36] The most distinctive hypochromic/achromic macules are known as “ash-leaves” because they resemble leaves and are tapering at one end and rounded at the other (Figure 2). [35]
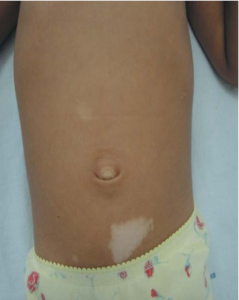
FIGURE 2: “Ash-Leaf” hypomelanotic macule
In 28% of the cases, they can also be rounded, or “confetti-like.” These could be one or more, 1-2 mm in diameter, and they would impact the distal portion of the limbs symmetrically all the way around (Figure 3). The “confetti-like” hypomelanosis is frequently misdiagnosed since ultraviolet light isn’t used regularly, which makes dyschromia easier to see. Achromic macules have a normal amount of melanocytes in a histological examination, but fewer, smaller, and less melanized melanosomes.
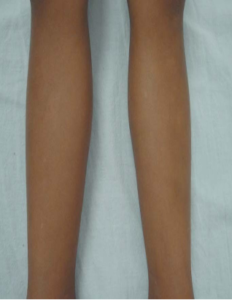
FIGURE 3: “Confetti-like” hypomelanotic macules on the anterior surface of legs
“Café-au-lait” macules may exist, although according to certain research, their frequency is comparable to that of the general population.[35] Skin-colored to violaceous papules, depending on which type of tissue has a higher proportion of fibrous or angiomatous tissue, are the characteristic of angiofibromas. Eighty percent of affected youngsters older than five years old have them.[3] They mostly affect the upper lip sparing the nasolabial folds, cheeks, and chin, bilaterally and symmetrically (Figures 4, 5 and 6). When they happen one-sidedly, mosaicism could result. [35-37]
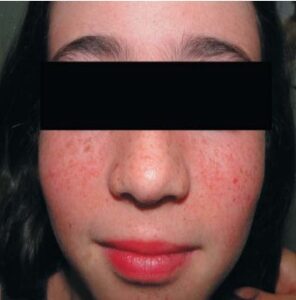
FIGURE 4: Facial angiofibromas at early stage
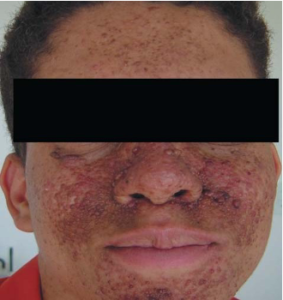
FIGURE 5: Facial angiofibromas
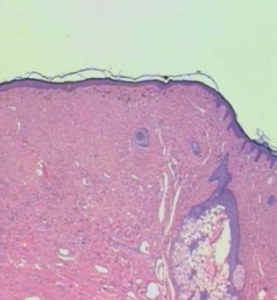
FIGURE 6: Histopathological examination of facial angiofibroma: HE. 25X
Excessive malar erythema is observed in clinical observations before the beginning of angiofibromas.[35] A skin-colored to brown fibrotic plaque known as a “shagreen patch” may have existed from birth, but it usually first manifests around the age of three (Figure 7).[35, 36]
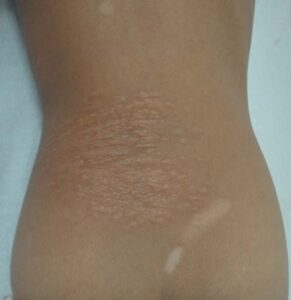
FIGURE 7: Shagreen patch
It frequently appears during puberty and affects 54% of children over the age of five. It almost always affects the lower dorsal area with many satellite papules.[3] skin-colored papules gathered together at the plaque’s distinctive location could be regarded as identical.[35, 36] Though less common, forehead fibrous plaques are thought to be pathognomonic for TSC. They may be skin-colored or brown, and they manifest as a unilateral plaque in the frontal region with a fibrotic aspect (Figure 8). [35]
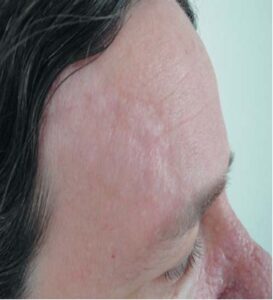
FIGURE 8: Fibrous forehead plaque
One of the last changes to appear in TSC is periungual fibroma, also known as Koenen’s tumor, and its incidence tends to rise with age (Figure 9).[37] Over time, tumors often recur and primarily affect women, particularly in the toe nails.
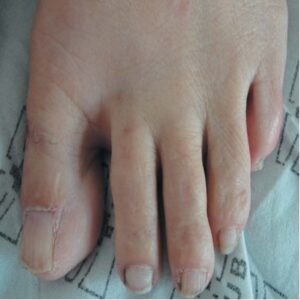
FIGURE 9: Koenen’s tumor
Sometimes the only indication of a tumor remnant left behind after the patient removed it is a longitudinal depression in the nail, which corresponds to a fibroma that is still present in the nail matrix (Figure 10). [35]
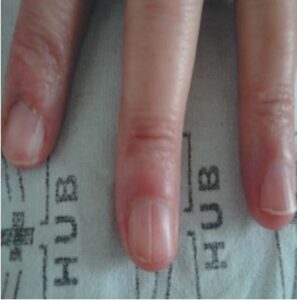
FIGURE 10: Longitudinal canalicular nail depression, corresponding to ungual fibroma in the nail matrix
Oral cavity
TSC may have an impact on the gums and teeth. Gingival fibromas are uncommon in children under the age of eleven and impact approximately 36% of people with TSC. They can cause flesh-colored or erythematous lesions and are typically found in the anterior gingiva. [35, 38]
Although enamel pits have been reported, they are frequently misdiagnosed during examinations and are not thought to be pathognomonic for TSC.[39] For this reason, a thorough evaluation of the oral cavity is essential, including a look at the gingiva behind dentures, to identify these alterations.
Renal manifestations
In TSC, renal alterations are the primary cause of morbidity and mortality. Renal angiomyolipomas, which manifest before adolescence and are present in 93% of cases, are the most prevalent type among them.[3] Typically, they are asymptomatic and bilateral. They may result in renal function loss and significant hematuria, which is more common in tumors bigger than 4 cm. Although they develop slowly, there are rare instances where their size might expand by up to 4 cm every two years. [35, 36]
The closeness of the TSC2 gene, which is situated on chromosome 16, and the polycystic kidney disease 1 (PKD1) gene results in concurrent polycystic kidney disease. The condition primarily affects children and causes a gradual decline of kidney function that can lead to chronic renal failure.[35, 36] The estimated incidence of renal cell carcinoma in patients with TSC is comparable to that of the general population. Unfortunately, the early development of these tumors increases the morbidity and death rate of TSC patients.[1]
Pulmonary manifestations
Because of the proliferation of smooth muscle, known as pulmonary lymphangioleiomyomatosis (LAM), and the replacement of alveolar tissue by many cysts, lung disease manifestation in TSC has a significant negative impact on the organ’s function. [40] In 1 to 3% of cases, it happens, ideally in women who are not yet menopausal. Males rarely get pulmonary involvement. The regulating effect of estrogen on cellular signaling pathways implicated in TSC and on the migration of TSC2 defective cells may account for this preference for females. Cough and increasing dyspnea are the main symptoms; hemoptysis and pneumothorax are also possible. In addition to being a therapeutic challenge, LAM has a progressive course that carries a cautious prognosis and has the potential to cause respiratory failure.[3]
Cardiac manifestations
The most frequent cardiac tumor in TSC is rhabdomyoma. It is the most common clinical alteration in fetuses and infants because it occurs earlier in life. [41] Depending on their size, quantity, and placement within the ventricles, they can cause cardiomegaly, cardiac murmurs, irregular blood flow, arrhythmias, non-immune hydrops fetalis, and even death. Because they are not stimulated by maternal hormones throughout the first few years of life, they typically regress entirely.[42]
Rhabdomyoma can cause a wide range of symptoms, however, the majority of cases have none at all.[41] In a recent study, foci of fat attenuation were discovered in 64% of the 55 individuals with TSC whose chest CT scans were reviewed by Adriaensen et al. The exact clinical relevance of these observations is still unknown, however, the authors speculate that they might constitute one of TSC’s hallmarks.[43]
Neurological manifestations
The three most prevalent neurological clinical symptoms associated with TSC are autism, cognitive delay, and epilepsy. [44] Children with TSC frequently experience seizures, with infantile spasms are thought to be the most frequently identified subtype during the first year of life. [36] Ninety percent of the cases may have epilepsy. Its more severe cognitive delay and refractoriness are linked to its early onset.[44] Reduced neural inhibition resulting from molecular changes in GABA receptors, which are found in large cells and dysplastic neurons, can account for epileptogenesis in TSC. Seizures may begin earlier and become more severe in response to GABAergic insufficiency. [3]
Following immunohistochemical analysis of surgical specimens from epileptic patients with TSC, overexpression of P-glycoprotein, encoded by the MDR1 gene, in the epileptogenic tissue, has been proposed as a possible explanation for refractory seizures in TSC. This glycoprotein would be elevated in these situations, impairing the therapeutic response, and it may be in charge of the clearance of antiepileptic medications.[45] Patients with TSC who had more than three interictal epileptogenic foci were found to have worse cognitive development in a study using serial analysis of their interictal electroencephalogram (EEG) records. Additionally, it was demonstrated that the frequency of interictal epileptogenic foci increased with the early onset of seizures. [46]
The most common epileptic encephalopathy in the first year of life is West syndrome, which is characterized by spasm-type seizures, delayed psychomotor development, and hypsarrhythmia in the electroencephalogram. TSC is the most common cause of West syndrome, following severe neonatal asphyxia and congenital brain malformations. This information is crucial for clinical practice. It is therefore necessary to detect both diseases, which are related because 30% of infants with TSC and epilepsy who are under a year old will go on to acquire West syndrome.[47]
Ninety to ninety-five percent of TSC cases have abnormal neuroimaging scans showing tubers in the cerebral cortex, subependymal nodules in the lateral ventricle walls, and subependymal giant cell tumors.[35] Numerous tubers, their bilateralism, and their location in the temporal lobe have all been linked to higher levels of neurological involvement in a series of investigations examining the association between imaging and clinical manifestations of TSC. [48, 49] Histopathological analysis reveals that the brain’s cortical tubers lack the typical laminated architecture. Large cells that resemble astrocytes are also present, however they test positive for glial and neuronal markers. Vascular stroma and cells resembling astrocytes are found in subependymal nodules.[35]
Other manifestations
In 50% of cases, calcified retinal astrocytic hamartoma may be present. [36, 50] They usually manifest bilaterally and start to show around age two. Most of the time, they have no symptoms, in which case the optic nerve or macula are unaffected. In addition, patients may exhibit hypochromic macules in the retina, which are similar to skin macules. [3]
Liver changes
In rare instances of TSC, several hepatic angiomyolipomas have been documented; given that they are typically asymptomatic, this incidence may be the consequence of underdiagnosis. Adult individuals with TSC have a greater frequency of hepatic hamartomas (23–45%) according to ultrasonographic examination. There is a little bias against women.[3]
Diagnosis
Clinical signs are the primary basis for the initial diagnosis of tuberous sclerosis. Children who have hypomelanotic macules and seizures should be evaluated for this disease. Clinical and radiological diagnostic criteria were changed in 2000 due to TSC’s clinical heterogeneity. Depending on the number of main and minor criteria that are met, the diagnosis can be considered definitive, probable, or feasible (Table 1). [36, 51]
| Minor criteria: | Major criteria: |
| Multiple pits in dental enamel | Facial angiofibromas or forehead plaques |
| Hamartomatous rectal polyps | Nontraumatic ungual or periungual fibroma |
| Bone cysts | Hypomelanotic macules (more than 3) |
| Cerebral white matter radial migration lines | Shagreen patch |
| Gingival fibromas | Multiple retinal nodular hamartomas |
| Nonrenal hamartomas | Cortical tubers |
| Retinal achromic patch | Subependymal nodules |
| ”Confetti-like” hypomelanotic macules | Subependymal giant cell astrocytoma |
| Multiple renal cysts | Cardiac rhabdomyoma (single or multiple) |
| Pulmonary lymphangioleiomyomatosis | |
| Renal angiomyolipoma |
- Definite diagnosis: two major criteria or one major and two minor criteria.
- Probable diagnosis: one major criterion and one minor criterion.
- Possible diagnosis: a major criterion or two or more minor criteria.
When one major and two minor criteria, or at least two major criteria, are present, the diagnosis is deemed definitive. When one main and one minor criterion are met, a probable diagnosis is made. The existence of one main criterion or two or more minor criteria is referred to as a possible diagnosis. [35, 36, 51]
When cerebral white matter radial migration and cortical dysplasia coexist, they will be regarded as a single criterion. Before validating the diagnosis, additional criteria for TSC must be met when LAM and renal angiomyolipomas develop simultaneously. Multiple renal cysts, nonrenal hamartomas, and hamartomatous rectal polyps should all be histologically proven. Bone cysts and cerebral white matter radial migration can be diagnosed only by imaging examination identification.[51] A few more tests should be carried out as soon as clinical signs of TSC appear to confirm the diagnosis and identify the underlying causes of the symptoms.
Therefore, before a cranial computed tomography (CT) or magnetic resonance imaging (MRI) can be ordered for a possible diagnosis of TSC, an electroencephalogram (EEG) must be ordered in the event of a seizure; renal ultrasound must be ordered for any suspected patient; women must request pulmonary function tests, echocardiography, electrocardiogram, ophthalmoscopy, and pulmonary CT; and children with polycystic kidneys and adults with extensive renal involvement must request renal function tests.
Assessments of neuropsychomotor development and behavior are also necessary to rule out the presence of mental retardation, hyperactivity, and autism. [36]
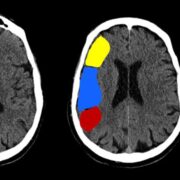
Rhabdomyomas can cause abnormalities on echocardiography even before neurological and cutaneous symptoms show up. [42] Subependymal nodules, frequently calcified, are visible on cranial CT scanning. These nodules are found in the lateral ventricle walls and may be extending into the ventricular chamber (Figure 11). [3, 35, 36] When evaluating cortical tubers, FLAIR sequence-based brain MRI performs better than other neuroimaging techniques (Figure 12). Using this technique, tubers could be linked to white matter anomalies like radial migration lines. [35, 36, 51] A fetal magnetic resonance imaging scan reveals changes from the 26th week of gestation, when the central nervous system contains tubers (Figure 13). [3]
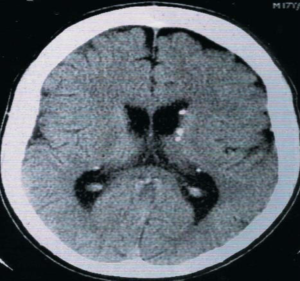
FIGURE 11: Cranial computed tomography scan: calcification in the ventricular walls.35
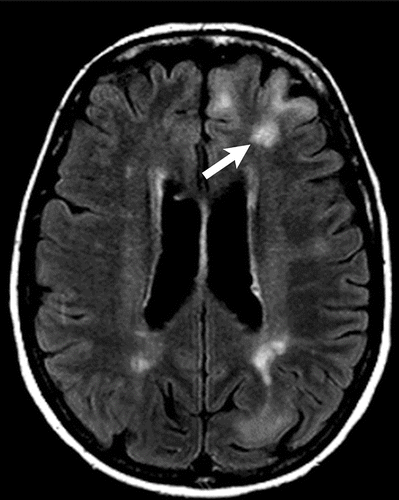
Figure 12. Axial FLAIR MR image shows multiple hyperintense lesions (arrow) in the cortex, consistent with cortical tubers in tuberous sclerosis.57
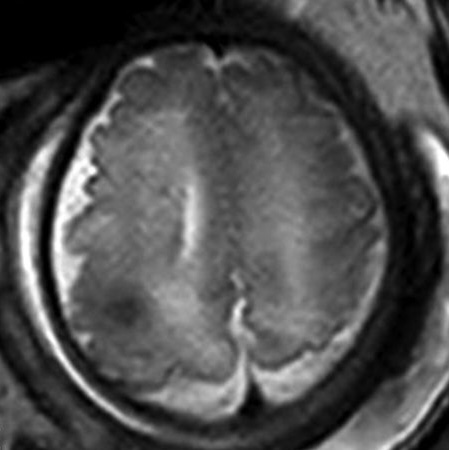
Figure 13: single-shot T2-weighted EPI shows cortical tuber.58
For confirmation and identification of the mutation causing TSC, a genetic analysis may be requested, even though it is not included in the traditional diagnostic criteria because of its limited availability. The evaluation and counseling of healthy relatives of individuals with TSC may also benefit from this test.
Monitoring
For children, a brain MRI or cranial CT scan should be done every one to three years; for adults, the frequency should be lower. When necessary, early detection of intracranial abnormalities allows for the full excision of malignancies. [36]
For women who have pulmonary LAM symptoms, a chest CT scan and pulmonary function tests are to be carried out every six to twelve months. [36] To track the development of renal angiomyolipomas, adolescents and adults should have renal ultrasonography performed every one to three years. [3, 36]
Genetic counselling
To develop early therapy for potential tumor consequences and to increase awareness of the potential transmissibility of TSC to future generations, it is crucial to actively look for cases of TSC in undiagnosed relatives who may have a less noticeable clinical manifestation. It is known that a patient with TSC has a 50% probability of carrying the illness to term, and that a healthy couple with a kid with TSC has a 2% chance of conceiving another child with the condition. [36]
Due to the delayed start of facial angiofibromas and ungual fibromas in certain situations, medical evaluation of healthy family members who intend to have children should involve cranial CT scanning, renal ultrasonography, and dermatological examination. [35]
As was already noted, genetic testing helps identify the mutations causing TSC instances and assesses alterations in healthy relatives to determine the likelihood that another child would be born with the illness.
Despite this, gonadal mosaicism is prevalent in around 2% of healthy parents of TSC children. Because these mutations would not be discovered via a genetic evaluation, the likelihood that the following child will experience TSC would still exist.[3]
Monitoring of patients with TSC is not usually recommended in the context of genetic evaluation. Molecular genetic testing aids doctors in clinical practice before an ambiguous clinical and radiological diagnosis, the requirement for family member evaluation, and prenatal diagnosis of the illness. The Tuberous Sclerosis Alliance states that polymerase chain reaction, DNA sequencing, and TSC1 and TSC2 dosage can all be used for genetic testing for the Tuberous Sclerosis Complex. [47]
Treatment
The goals of TSC treatment are to manage the symptoms brought on by hamartomas and take preventative steps to keep the affected organ from losing its functionality. It requires a comprehensive approach because it is a systemic condition. Recurrence of facial angiofibromas is common and can be treated with dermabrasion, surgical excision, electrocautery, and argon laser therapy. [35, 36] The best treatment for fibrous tumors is CO2 laser therapy.[36]
Surgery may not be performed on face tumors until late adolescence, when their growth is at its fastest, due to their recurrent and progressive growth. [35] Ungual fibromas that cause pain or bleeding often can be surgically removed and treated with argon laser therapy or electrocautery, although recurrence is not unusual. [35, 36]
Arterial embolization is the recommended treatment for renal angiomyolipomas that result in persistent hematuria. Since tumors larger than 4 cm are the most likely to develop severe hematuria, close monitoring is necessary, and the need for prophylactic embolization should be evaluated.[36] When surgical therapy entails significant dangers, this operation should also be carried out for tumors located in the center of the body. Angiogenesis inhibitors may be used to enhance the prognosis of TSC and stop the growth of angiomyolipomas.[3] If cardiac rhabdomyomas are symptomatic, they should be surgically removed. Clinical care is provided for cardiac arrhythmias.[36]
Patients with epilepsy should take anticonvulsants; vigabatrin is the most effective medication. It is advised to utilize corticotropin or prednisolone for infantile spasms. [36] Surgically removing epileptogenic foci has reduced medication resistance in patients with TSC and refractory epilepsy by 57%. Behavioral issues and cognitive function decline can be lessened with surgery.[36] Radiotherapy does not always produce a favorable response in cases of subependymal astrocytoma, and it also increases the incidence of glioblastomas. Therefore, surgical resection is the preferable treatment in this scenario. [36]
Following the revelation that the mTOR pathway’s regulation is linked to the growth of tumors in TSC, several intriguing studies have drawn a lot of interest. The mTOR pathway is influenced by interferon-gamma and -alpha, which deactivates 4E-BP1, which may be highly beneficial in the therapy of TSC.[3]
mTOR pathway inhibitors, like sirolimus, sometimes referred to as rapamycin, exhibit antiproliferative and immunosuppressive properties. They are in charge of restoring this pathway’s proper operation in cells lacking TSC1 or TSC2. In patients with TSC, such as those with renal angiomyolipoma, subependymal giant cell astrocytoma, and sporadic LAM, this medication is useful in decreasing the volume of tumors.
Individuals with TSC2 mutations demonstrated improved learning, while those with LAM demonstrated rapamycin-induced recovery of lung function. Even though the medication had a decent therapeutic impact in treating renal angiomyolipomas, there was a resumption of uncontrolled tumor growth following drug withdrawal. [52]
In a clinical trial with psoriasis patients, topical therapy with rapamycin was demonstrated to be safe and feasible. [53] Given the potential for fewer adverse effects and a stronger effect on the skin, the actual function of this mode of administration is encouraging. But as of yet, there isn’t any reliable scientific proof. [54]
It is thought that gene therapy will eventually allow for the treatment of mutations that cause TSC by either reintroducing the lost gene, which will allow the cell to exhibit a normal phenotype and prevent the clinical changes associated with TSC, or reintroducing the defective gene, which will cause the mutated cells to undergo apoptosis.[55, 56]
References
[Read more]
- Orlova, K.A. and P.B. Crino, The tuberous sclerosis complex. Annals of the New York Academy of Sciences, 2010. 1184(1): p. 87-105.
- Callen, J.P., et al., Dermatological Signs of Internal Disease E-Book: Expert Consult-Online and Print. 2009: Elsevier Health Sciences.
- Curatolo, P., R. Bombardieri, and S. Jozwiak, Tuberous sclerosis. The Lancet, 2008. 372(9639): p. 657-668.
- Lendvay, T.S. and F.F. Marshall, The tuberous sclerosis complex and its highly variable manifestations. The Journal of urology, 2003. 169(5): p. 1635-1642.
- Ozcan, U., et al., Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Molecular cell, 2008. 29(5): p. 541-551.
- Napolioni, V. and P. Curatolo, Genetics and molecular biology of tuberous sclerosis complex. Current Genomics, 2008. 9(7): p. 475-487.
- Roach, E., et al., Tuberous sclerosis consensus conference: recommendations for diagnostic evaluation. Journal of child neurology, 1999. 14(6): p. 401-407.
- Devlin, L.A., et al., Tuberous sclerosis complex: clinical features, diagnosis, and prevalence within Northern Ireland. Developmental medicine and child neurology, 2006. 48(6): p. 495-499.
- Wolff, K., et al., Fitzpatrick’s dermatology in general medicine. 2008: McGraw-Hill New York.
- O’Callaghan, F.J., et al., An epidemiological study of renal pathology in tuberous sclerosis complex. BJU international, 2004. 94(6): p. 853-857.
- Kwiatkowski, D., Tuberous sclerosis: from tubers to mTOR. Annals of human genetics, 2003. 67(1): p. 87-96.
- Yates, J.R., et al., Female germline mosaicism in tuberous sclerosis confirmed by molecular genetic analysis. Human molecular genetics, 1997. 6(13): p. 2265-2269.
- Rose, V.M., et al., Germ-line mosaicism in tuberous sclerosis: how common? The American Journal of Human Genetics, 1999. 64(4): p. 986-992.
- Dabora, S.L., et al., Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. The American Journal of Human Genetics, 2001. 68(1): p. 64-80.
- Jones, A.C., et al., Comprehensive mutation analysis of TSC1 and TSC2—and phenotypic correlations in 150 families with tuberous sclerosis. The American Journal of Human Genetics, 1999. 64(5): p. 1305-1315.
- Sancak, O., et al., Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. European Journal of Human Genetics, 2005. 13(6): p. 731-741.
- Au, K.-S., et al., Complete inactivation of the TSC2 gene leads to formation of hamartomas. The American Journal of Human Genetics, 1999. 65(6): p. 1790-1794.
- Green, A.J., M. Smith, and J.R. Yates, Loss of heterozygosity on chromosome 16p13. 3 in hamartomas from tuberous sclerosis patients. Nature genetics, 1994. 6(2): p. 193-196.
- Henske, E.P., et al., Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. American journal of human genetics, 1996. 59(2): p. 400.
- Han, S., et al., Phosphorylation of tuberin as a novel mechanism for somatic inactivation of the tuberous sclerosis complex proteins in brain lesions. Cancer research, 2004. 64(3): p. 812-816.
- Tee, A.R., et al., Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Current biology, 2003. 13(15): p. 1259-1268.
- Nellist, M., M. Goedbloed, and D. Halley, Regulation of tuberous sclerosis complex (TSC) function by 14-3-3 proteins. Biochemical Society Transactions, 2003. 31(3): p. 587-591.
- Fingar, D.C., et al., Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes & development, 2002. 16(12): p. 1472-1487.
- Garami, A., et al., Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Molecular cell, 2003. 11(6): p. 1457-1466.
- Saucedo, L.J., et al., Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nature cell biology, 2003. 5(6): p. 566-571.
- Inoki, K., M.N. Corradetti, and K.-L. Guan, Dysregulation of the TSC-mTOR pathway in human disease. Nature genetics, 2005. 37(1): p. 19-24.
- Kwiatkowski, D.J. and B.D. Manning, Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Human molecular genetics, 2005. 14(suppl_2): p. R251-R258.
- Sarbassov, D.D., S.M. Ali, and D.M. Sabatini, Growing roles for the mTOR pathway. Current opinion in cell biology, 2005. 17(6): p. 596-603.
- Dann, S.G. and G. Thomas, The amino acid sensitive TOR pathway from yeast to mammals. FEBS letters, 2006. 580(12): p. 2821-2829.
- Rosner, M., A. Freilinger, and M. Hengstschläger, Akt regulates nuclear/cytoplasmic localization of tuberin. Oncogene, 2007. 26(4): p. 521-531.
- Gómez, M.R., History of the tuberous sclerosis complex. Brain and Development, 1995. 17: p. 55-57.
- Qian, F., et al., The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell, 1996. 87(6): p. 979-987.
- Brasier, J.L. and E.P. Henske, Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. The Journal of clinical investigation, 1997. 99(2): p. 194-199.
- Stillwell, T.J., M.R. Gomez, and P.P. Kelalis, Renal lesions in tuberous sclerosis. The Journal of urology, 1987. 138(3): p. 477-481.
- Webb, D., et al., The cutaneous features of tuberous sclerosis: a population study. British Journal of Dermatology, 1996. 135(1): p. 1-5.
- Yates, J.R., et al., The Tuberous Sclerosis 2000 Study: presentation, initial assessments and implications for diagnosis and management. Archives of disease in childhood, 2011. 96(11): p. 1020-1025.
- Aldrich, C.S.L., et al., Acral lesions in tuberous sclerosis complex: insights into pathogenesis. Journal of the American Academy of Dermatology, 2010. 63(2): p. 244-251.
- Júnior, H.M., et al., Oral manifestations leading to the diagnosis of familial tuberous sclerosis. Indian Journal of Dental Research, 2010. 21(1): p. 138-140.
- marinho Pereira, A.L., Esclerose tuberosa: relato de caso. Anais brasileiros de dermatologia, 1998. 73(5): p. 431-434.
- Park, H.Y., et al., A nationwide survey of lymphangioleiomyomatosis in Korea: recent increase in newly diagnosed patients. Journal of Korean medical science, 2010. 25(8): p. 1182-1186.
- Quek, S., et al., Cardiac manifestations in tuberous sclerosis: A 10‐year review. Journal of paediatrics and child health, 1998. 34(3): p. 283-287.
- Yinon, Y., et al., Fetal cardiac tumors: a single‐center experience of 40 cases. Prenatal diagnosis, 2010. 30(10): p. 941-949.
- Adriaensen, M.E., et al., Fatty foci in the myocardium in patients with tuberous sclerosis complex: common finding at CT. Radiology, 2009. 253(2): p. 359-363.
- Samir, H., H.A. Ghaffar, and M. Nasr, Seizures and intellectual outcome: clinico-radiological study of 30 Egyptian cases of tuberous sclerosis complex. european journal of paediatric neurology, 2011. 15(2): p. 131-137.
- Lazarowski, A., et al., Multidrug resistance proteins in tuberous sclerosis and refractory epilepsy. Pediatric neurology, 2004. 30(2): p. 102-106.
- Jansen, F.E., et al., Consistent localization of interictal epileptiform activity on EEGs of patients with tuberous sclerosis complex. Epilepsia, 2005. 46(3): p. 415-419.
- Rodrigues, D.A., C.M. Gomes, and I.M.C. Costa, Tuberous sclerosis complex. Anais brasileiros de dermatologia, 2012. 87: p. 184-196.
- Chou, I.-J., et al., Neuroimaging correlation with neurological severity in tuberous sclerosis complex. European Journal of Paediatric Neurology, 2008. 12(2): p. 108-112.
- Zaroff, C.M., et al., Mental retardation and relation to seizure and tuber burden in tuberous sclerosis complex. Seizure, 2006. 15(7): p. 558-562.
- Rosser, T., A. Panigrahy, and W. McClintock. The diverse clinical manifestations of tuberous sclerosis complex: a review. in Seminars in pediatric neurology. 2006. Elsevier.
- Hyman, M.H. and V.H. Whittemore, National Institutes of Health consensus conference: tuberous sclerosis complex. Archives of neurology, 2000. 57(5): p. 662-665.
- Bissler, J.J., et al., Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. New England Journal of Medicine, 2008. 358(2): p. 140-151.
- Ormerod, A., et al., Treatment of psoriasis with topical sirolimus: preclinical development and a randomized, double‐blind trial. British Journal of Dermatology, 2005. 152(4): p. 758-764.
- Haemel, A.K., A.L. O’Brian, and J.M. Teng, Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Archives of dermatology, 2010. 146(7): p. 715-718.
- Ward, L.S., Entendendo o processo molecular da tumorigênese. Arquivos Brasileiros de Endocrinologia & Metabologia, 2002. 46: p. 351-360.
- Mamane, Y., et al., mTOR, translation initiation and cancer. Oncogene, 2006. 25(48): p. 6416-6422.
-
Wang MX, Segaran N, Bhalla S, Pickhardt PJ, Lubner MG, Katabathina VS, Ganeshan D. Tuberous Sclerosis: Current Update. Radiographics. 2021 Nov-Dec;41(7):1992-2010. doi: 10.1148/rg.2021210103. Epub 2021 Sep 17. PMID: 34534018.
- Goergen SK, Fahey MC. Prenatal MR Imaging Phenotype of Fetuses with Tuberous Sclerosis: An Institutional Case Series and Literature Review. AJNR Am J Neuroradiol. 2022 Apr;43(4):633-638. doi: 10.3174/ajnr.A7455. Epub 2022 Mar 24. PMID: 35332020; PMCID: PMC8993194.
[\read]