Article Title: Parkinson’s Disease
Author: Farah Moh’d Sbeih Fuad Awayes.
Editor: Dr Ethar Hazaimeh
Keywords: Parkinson’s disease, Parkinsonism, Lewy bodies.
Overview:
Parkinson’s disease (PD) is the most common chronic, progressive movement disorder of the central nervous system and is the second most common neurodegenerative disease after Alzheimer’s disease. [1][2] It involves the degeneration of the dopaminergic neurons in the substantia nigra pars compacta (SNpc) where it manifests mostly as non-motor symptoms that precedes the classical motor symptoms. [3] The motor symptoms usually appear after the degeneration of 50-70% of the SNpc neurons. [4]
PD is named after the British physician James Parkinson who wrote “An Essay on the Shaking Palsy” in 1817 and described it as a neurological syndrome. Later, in 1872, Jean-Martin Charcot suggested to name it differently to differentiate it from other disorders, especially multiple sclerosis, and since then it was called Parkinson’s disease. [5][6][7]
Epidemiology:
1-2 per 1000 are affected at any time; [2] studies recognized an increase in PD’s incidence with increasing age occupying 1% of people above 65 years of age. [8] Also, it was noticed that women have lower incidence rate and higher age at which symptoms would appear as there may be a protective factor from female sex hormones. [9] Men are twice more prone to PD than women and it was found that Hispanics have the highest susceptibility in US. [10]
Pathogenesis:
PD revolves around the combination of neurons degeneration in the SNpc and pigment loss. Another crucial hallmark is the falsely folded α-synuclein protein following a mutation in the SNCA gene [11]; when misfolded, they become insoluble and accumulate intracellularly to form what we call Lewy bodies. Lewy bodies can also be found in the spinal cord and peripheral nervous system, including the vagus nerve, salivary glands, sympathetic ganglia, cardiac plexus, enteric nervous system, adrenal medulla, and sciatic nerve. [12]
Etiology:
The cause of PD remains unknown, yet, it is believed to be multifactorial. Genetics contribute to 5% of PD cases; six main genes are responsible for the autosomal dominant forms of Parkinson’s disease which are SNCA, LRRK2, VPS35, EIF4G1, DNAJC13 and CHCHD2. [3] LRRK2 mutations account for 4% of familial PD and 1% of sporadic PD and is considered the most common PD-causing mutation. [13] LRRK2 normally encodes the leucine-rich repeat kinase 2, a large multidomain protein involved in various cellular processes and has a part in the innate immunity, therefore, a mutation in LRRK2 will cause a Gly2019Ser amino acid substitution, thus, increasing the kinase activity. [14][15]
Environment plays an important role as well; infections, MPTP, heavy metals, pesticides and herbicides exposure, living in rural areas, head trauma, drinking well water and others are some examples. MPTP exposure has been specifically linked to PD as it crosses the blood brain barrier and is then taken up by dopaminergic cells, hence, destroying them after being converted to MPP+ by astrocytes. [16][17] Strangely, cigarette smoking and caffeine intake have been inversely linked to PD. [18][19]
Clinical Presentation:
Progression of early PD undergoes three phases according to The Movement Disorder Task Force. First, the preclinical phase, where patients have no clinical symptoms but neurodegeneration occurs. Second, the prodromal phase, where symptoms, often non-motor symptoms, begin to appear but are not PD-specific and are inadequate for diagnosis. Third, the clinical phase, where symptoms are obvious enough for PD diagnosis. [20][21][22]
The classical motor symptoms arise unilaterally and asymmetrically; there are four cardinal symptoms in PD and these are tremor, bradykinesia or akinesia, muscular rigidity and postural instability. [23]
- Tremor is usually the first motor symptom to appear and it is defined as an involuntary continuous shaking when muscles are at rest or relaxed; although most PD patients develop resting tremor, 50% also experience tremor with extended arms after latency. [20] It is characterized by supination and pronation, or pill-rolling.
- Bradykinesia is the decline in voluntary or spontaneous movement while akinesia means loss of movement. [24]
- Rigidity means muscle stiffness and difficulty in motion when moved passively.
- Postural instability is the inability to maintain an upright, balanced position; it usually appears later in the disease course and it is considered the least responsive to levodopa therapy and the most threatening symptom contributing to increased falls, hip fractures, and dependency. [25]
The course of progression can be seen in Figure 1.

Figure 1: Clinical symptoms and time course of PD progression. [3] EDS = Excessive Daytime Sleepiness. MCI = Mild Cognitive Impairment. RBD = REM sleep Behavior Disorder.
As mentioned earlier, non-motor symptoms precede motor symptoms in most cases, and can last up to 20 years or more; these symptoms include:
- Autonomic symptoms like orthostatic hypotension, constipation, faecal incontinence, nausea and vomiting. [25]
- Neuropsychiatric symptoms including depression, hallucinations, psychosis and panic attacks. [25]
- Cognitive symptoms like dementia and memory loss. [25]
- Sleep disturbances including excessive daytime sleepiness (EDS) and rapid eye movement sleep behavior disorder (RBD). [25]
- Sensory abnormalities like pain, numbness and anosmia. [25]
There are also the premotor symptoms which are RBD, constipation, depression and anosmia too. [26][27]
Work up and Diagnosis:
Diagnosis of PD is quite challenging especially in the early stages since it has overlapping signs and symptoms with other parkinsonian syndromes. The definite diagnosis is post-mortem when autopsied in 76% of cases. [28]
Clinical diagnosis relies mostly on history and neuropathological assessment to identify the presence of the cardinal motor signs; it can also be assisted by response to levodopa therapy also called the levodopa test. [29]
In UK, they improved the diagnostic sensitivity and specificity to 98.6% and 91.1% respectively with the use of the UK Parkinson’s Disease Society Brain Bank (UKPDSBB) clinical criteria shown in Figure 2. [23]

Figure 2: UK Parkinson’s Disease Society Brain Bank criteria [23]
Furthermore, disease staging was established by Braak and colleagues to reflect the location of lewy bodies deposition in different brain parts, this guides in determining the disease progression; only 10-20% of cases are exceptions that opposed this staging scheme. [30]
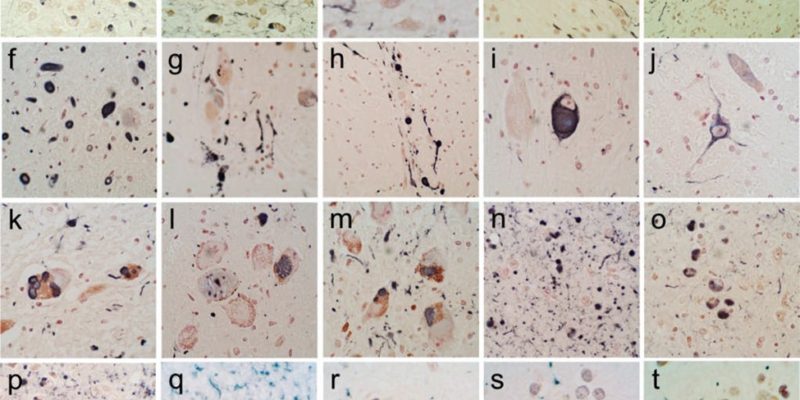
Stage 1 in which lewy bodies begin to deposit in the olfactory bulb and/or the glossopharyngeal and vagal nerves, followed by stage 2 where they develop in the medulla oblongata and the pontine tegmentum. Stage 3, when motor symptoms start to show, is when the α-synuclein inclusions begin to appear in the amygdala and the substantia nigra. Stage 4, lewy bodies reach the temporal cortex. Finally, stages 5 and 6 where cognitive alterations are noticed when it reaches the neocortex. [31] Figure 3 shows immunohistochemical staining for α-synuclein in different brain regions.

Figure 3: Immunohistochemical staining for α-synuclein in different brain regions. +ve immunostaining is black; the counterstain is neutral red. (a–e) The olfactory bulb & tract. An enlarged, abnormal neurite within the olfactory tract shown in (d). +ve fibers coursing in parallel array through the internal plexiform layer shown in (e). (f–j) The anterior medulla. The dorsal motor nucleus of the vagus nerve is shown in (f), the raphe nucleus in (g) and (i), the internal tract of the glossopharyngeal nerve in (h) and the lateral reticular nucleus in (j). (k–m) The locus ceruleus in the pons. (n–p) The amgydala. (q) The cingulate gyrus. (r) The middle temporal gyrus. (s) The middle frontal gyrus. (t) The inferior parietal lobule. [32]
Moreover, imaging aids in confirming the diagnosis and further understanding of the pathophysiology. Positron Emission Tomography (PET) or Single Photon Emission Computed Tomography (SPECT) measures loss of SNpc dopaminergic nerves and provides a biochemical marker for disease progression. [33]
Differential Diagnosis:
Due to the complexity of PD diagnosis, misdiagnosis have been found in almost 25% of patients. The differential diagnosis of Parkinson disease includes:
- Essential tremor is either an action tremor when doing simple tasks or a postural tremor that arises bilaterally and symmetrically in most cases with absent bradykinesia, rigidity and postural instability; misdiagnosis occur when essential tremor is unilateral or is a resting tremor. [34][35]
- Progressive Supranuclear Palsy or Steel Richardson-Olszewski syndrome is a multisystem degenerative disorder; gaze palsy is the typical clinical picture that distinguishes it from PD easily; confusion arises in the early stages when symptoms overlap. [36]
- Multisystem Atrophy is a multisystem degenerative disorder that involves α-synuclein accumulation in the CNS with different degrees of autonomic involvement. The parkinsonian variant, or the MSA-p, is usually misdiagnosed with PD. [37]
- Dementia with Lewy bodies is a degenerative dementia that affects cognition, attention and causes visual hallucinations and parkinsonian signs that makes it difficult to differentiate. [38]
- Drug-induced parkinsonism is the most common secondary parkinsonism that may result in confusion at diagnosis especially with patients that have an unclear drug history. Various anti-dopaminergic drugs, mainly anti-psychotics and anti-emetics, induce this secondary parkinsonism. [39]
Management
There is no definite treatment for PD as all available options work as symptomatic relievers.
- Levodopa/Carbidopa remain the gold standard therapy; Levodopa is transported to the brain and is then converted to dopamine as a striatal dopamine substitute while Carbidopa prevents its breakdown peripherally and decreases nausea. [40] Chronic use of levodopa causes dyskinesia and motor fluctuation.
- Dopamine agonists can also be used such as Apomorphine that has both D1 and D2 receptor activity and same potency as Levodopa with less motor complications. Other Dopamine agonists include pramipexole, ropinirole and rotigotine. [41][42] Common side-effects of dopamine agonists and less commonly Levodopa include: nausea, hypotension, oedema, hallucinations, impulse control disorders and sudden sleep attacks.
- Catechol-O-Methyl Transferase (COMT) inhibitors including Entacapone and Tolcapone.
- Monoamine Oxidase Type B (MAO-B)inhibitors, like Selegiline and Rasagiline as they increase dopamine synaptic concentration.
COMT inhibitors and MAO-B inhibitors are both used along Levodopa to increase its half-life and bioavailability. [33][35]
- Amantadine, a N-Methyl-D-aspartate (NMDA) receptor antagonist, is used for levodopa-induced dyskinesia. [45]
- Cholinesterase inhibitor such as Rivastigmine is used in dementia caused by PD. [46] Autonomic dysfunction is treated mainly with mineralocorticoid and fludrocortisone; Droxidopa is used with orthostatic hypotension. [45][47] Depression is treated mainly with Selective Serotonin Reuptake Inhibitor (SSRI) since Tricyclic antidepressants can worsen orthostatic hypotension. [48]
Surgical intervention is used with severe motor complications and dyskinesias that do not respond well to medications; Deep Brain Stimulation (DBS) mimics ablative surgery by implanting a pacemaker-like device that generates electrical impulses targeting either the subthalamic nucleus, the thalamus’ ventral intermediate nucleus or the globus pallidus interna. [49] Patients who do not respond to dopaminergic therapy do not typically respond to DBS, therefore, to obtain maximal results, patients must be carefully selected preoperatively and implantation must be done accurately. [50] There are also side effects for DBS but they are found to be reversible; they include paraesthesia, ataxia, dysarthria, and frequent mood changes. [25] DBS electrodes are shown in CT images in Figure 4.

Figure 4: CT images show bilateral deep brain stimulation electrodes extending to the region of the subthalamic nuclei. Stimulator is housed on the chest wall. Case courtesy of Dr. Bruno Di Muzio [51]
Risk Factors:
Many risk factors were mentioned throughout this paper; to summarize the main factors they include:
- Age is the main risk factor. Incidence and risk increase with age.
- Males are more susceptible than females (2:1).
- Race/ethnicity. In US, Hispanics had the highest risk followed by Whites, Asians, and Blacks. [10]
- Genetics, mostly SNCA and
- Environmental factors like exposure to pesticides, heavy metals and others with dietary intake of free radical inducers such as iron and fat.
Prevention:
Studies proved that regular exercise including aerobic exercises with balance training and moderate intake of caffeine can play a role in diminishing the risk of developing PD. [52][19] Another study stated that the use of non-steroidal anti-inflammatory drugs (NSAIDs) can also reduce PD risk. [53] Moreover, diet has a part in prevention as well such as consuming antioxidants like vitamin E can neutralize the free radicals, thus, decreasing the oxidative damage [54]; on the other hand, dietary fat and iron induce free radicals and increase risk of PD. [55][56]
Prognosis
PD itself is not fatal but the complications that come along with it may decrease life expectancy. These complications include increased falls and trauma due to postural instability, recurrent infections and choking due to swallowing problems. [57][58] It was also noticed that developing dementia and the onset of which a patient does contributes to a poor outcome. [57]
References:
[1] M. T. Hayes, “Parkinson’s Disease and Parkinsonism.,” Am. J. Med., vol. 132, no. 7, pp. 802–807, Jul. 2019, doi: 10.1016/j.amjmed.2019.03.001.
[2] O.-B. Tysnes and A. Storstein, “Epidemiology of Parkinson’s disease.,” J. Neural Transm., vol. 124, no. 8, pp. 901–905, Aug. 2017, doi: 10.1007/s00702-017-1686-y.
[3] L. V Kalia and A. E. Lang, “Parkinson’s disease.,” Lancet (London, England), vol. 386, no. 9996, pp. 896–912, Aug. 2015, doi: 10.1016/S0140-6736(14)61393-3.
[4] P. K. Morrish, J. S. Rakshi, D. L. Bailey, G. V Sawle, and D. J. Brooks, “Measuring the rate of progression and estimating the preclinical period of Parkinson’s disease with [18F]dopa PET.,” J. Neurol. Neurosurg. Psychiatry, vol. 64, no. 3, pp. 314–319, Mar. 1998, doi: 10.1136/jnnp.64.3.314.
[5] J. Parkinson, “An Essay on the Shaking Palsy,” Whittingham Rowl. Sherwood, Needly Jones, 1817.
[6] C. J-M, De la paralysie agitante. 1872.
[7] C. G. Goetz, “The history of Parkinson’s disease: early clinical descriptions and neurological therapies.,” Cold Spring Harb. Perspect. Med., vol. 1, no. 1, p. a008862, Sep. 2011, doi: 10.1101/cshperspect.a008862.
[8] C. M. Tanner and S. M. Goldman, “Epidemiology of Parkinson’s disease.,” Neurol. Clin., vol. 14, no. 2, pp. 317–335, May 1996, doi: 10.1016/S0733-8619(05)70259-0.
[9] C. A. Haaxma et al., “Gender differences in Parkinson’s disease.,” J. Neurol. Neurosurg. Psychiatry, vol. 78, no. 8, pp. 819–824, Aug. 2007, doi: 10.1136/jnnp.2006.103788.
[10] S. K. Van Den Eeden et al., “Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity.,” Am. J. Epidemiol., vol. 157, no. 11, pp. 1015–1022, Jun. 2003, doi: 10.1093/aje/kwg068.
[11] M. H. Polymeropoulos et al., “Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease.,” Science, vol. 276, no. 5321, pp. 2045–2047, Jun. 1997, doi: 10.1126/science.276.5321.2045.
[12] D. G. Beach, T. G., Adler, C. H., Sue, L. I., Vedders, L., Lue, L., Walker, “Multi-organ distribution of phosphorylated a-synuclein histopathology in subjects with Lewy body disorders,” Acta Neuropathol, vol. 119, no. 6, pp. 689–702, 2010.
[13] D. G. Healy et al., “Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study.,” Lancet. Neurol., vol. 7, no. 7, pp. 583–590, Jul. 2008, doi: 10.1016/S1474-4422(08)70117-0.
[14] M. R. Cookson, “Cellular effects of LRRK2 mutations.,” Biochem. Soc. Trans., vol. 40, no. 5, pp. 1070–1073, Oct. 2012, doi: 10.1042/BST20120165.
[15] N. Dzamko and G. M. Halliday, “An emerging role for LRRK2 in the immune system.,” Biochem. Soc. Trans., vol. 40, no. 5, pp. 1134–1139, Oct. 2012, doi: 10.1042/BST20120119.
[16] J. W. Langston, P. Ballard, J. W. Tetrud, and I. Irwin, “Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis.,” Science, vol. 219, no. 4587, pp. 979–980, Feb. 1983, doi: 10.1126/science.6823561.
[17] B. R. Ransom, D. M. Kunis, I. Irwin, and J. W. Langston, “Astrocytes convert the parkinsonism inducing neurotoxin, MPTP, to its active metabolite, MPP+.,” Neurosci. Lett., vol. 75, no. 3, pp. 323–328, Apr. 1987, doi: 10.1016/0304-3940(87)90543-x.
[18] D. M. Morens, A. Grandinetti, D. Reed, L. R. White, and G. W. Ross, “Cigarette smoking and protection from Parkinson’s disease: false association or etiologic clue?,” Neurology, vol. 45, no. 6, pp. 1041–1051, Jun. 1995, doi: 10.1212/wnl.45.6.1041.
[19] G. W. Ross et al., “Association of coffee and caffeine intake with the risk of Parkinson disease.,” JAMA, vol. 283, no. 20, pp. 2674–2679, May 2000, doi: 10.1001/jama.283.20.2674.
[20] J. A. Obeso et al., “Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy.,” Mov. Disord., vol. 32, no. 9, pp. 1264–1310, Sep. 2017, doi: 10.1002/mds.27115.
[21] D. Berg et al., “Changing the research criteria for the diagnosis of Parkinson’s disease: obstacles and opportunities.,” Lancet. Neurol., vol. 12, no. 5, pp. 514–524, May 2013, doi: 10.1016/S1474-4422(13)70047-4.
[22] M. B. Stern, A. Lang, and W. Poewe, “Toward a redefinition of Parkinson’s disease.,” Mov. Disord., vol. 27, no. 1, pp. 54–60, Jan. 2012, doi: 10.1002/mds.24051.
[23] A. J. Hughes, S. E. Daniel, L. Kilford, and A. J. Lees, “Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases.,” J. Neurol. Neurosurg. Psychiatry, vol. 55, no. 3, pp. 181–184, Mar. 1992, doi: 10.1136/jnnp.55.3.181.
[24] A. Berardelli, J. C. Rothwell, P. D. Thompson, and M. Hallett, “Pathophysiology of bradykinesia in Parkinson’s disease.,” Brain, vol. 124, no. Pt 11, pp. 2131–2146, Nov. 2001, doi: 10.1093/brain/124.11.2131.
[25] T. A. Zesiewicz, “Parkinson Disease.,” Continuum (Minneap. Minn)., vol. 25, no. 4, pp. 896–918, Aug. 2019, doi: 10.1212/CON.0000000000000764.
[26] R. B. Postuma et al., “Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease.,” Mov. Disord., vol. 27, no. 5, pp. 617–626, Apr. 2012, doi: 10.1002/mds.24996.
[27] T. K. Khoo et al., “The spectrum of nonmotor symptoms in early Parkinson disease.,” Neurology, vol. 80, no. 3, pp. 276–281, Jan. 2013, doi: 10.1212/WNL.0b013e31827deb74.
[28] E. Tolosa, G. Wenning, and W. Poewe, “The diagnosis of Parkinson’s disease.,” Lancet. Neurol., vol. 5, no. 1, pp. 75–86, Jan. 2006, doi: 10.1016/S1474-4422(05)70285-4.
[29] C. D. Ward and W. R. Gibb, “Research diagnostic criteria for Parkinson’s disease.,” Adv. Neurol., vol. 53, pp. 245–249, 1990.
[30] H. Braak et al., “Pathology associated with sporadic Parkinson’s disease–where does it end?,” J. Neural Transm. Suppl., no. 70, pp. 89–97, 2006, doi: 10.1007/978-3-211-45295-0_15.
[31] H. Braak, K. Del Tredici, U. Rüb, R. A. I. de Vos, E. N. H. Jansen Steur, and E. Braak, “Staging of brain pathology related to sporadic Parkinson’s disease.,” Neurobiol. Aging, vol. 24, no. 2, pp. 197–211, 2003, doi: 10.1016/s0197-4580(02)00065-9.
[32] T. G. Beach et al., “Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction.,” Acta Neuropathol., vol. 117, no. 6, pp. 613–634, Jun. 2009, doi: 10.1007/s00401-009-0538-8.
[33] D. J. Brooks and N. Pavese, “Imaging biomarkers in Parkinson’s disease.,” Prog. Neurobiol., vol. 95, no. 4, pp. 614–628, Dec. 2011, doi: 10.1016/j.pneurobio.2011.08.009.
[34] O. Cohen, S. Pullman, E. Jurewicz, D. Watner, and E. D. Louis, “Rest Tremor in Patients With Essential Tremor: Prevalence, Clinical Correlates, and Electrophysiologic Characteristics,” Arch. Neurol., vol. 60, no. 3, pp. 405–410, Mar. 2003, doi: 10.1001/archneur.60.3.405.
[35] E. D. Louis, G. Levy, L. J. Côte, H. Mejia, S. Fahn, and K. Marder, “Clinical correlates of action tremor in Parkinson disease.,” Arch. Neurol., vol. 58, no. 10, pp. 1630–1634, Oct. 2001, doi: 10.1001/archneur.58.10.1630.
[36] E. Tolosa, F. Valldeoriola, and F. Cruz-Sánchez, “Progressive supranuclear palsy: clinical and pathological diagnosis,” Eur. J. Neurol., vol. 2, no. 4, pp. 259–273, 1995, doi: https://doi.org/10.1111/j.1468-1331.1995.tb00129.x.
[37] S. Gilman et al., “Consensus statement on the diagnosis of multiple system atrophy.,” J. Neurol. Sci., vol. 163, no. 1, pp. 94–98, Feb. 1999, doi: 10.1016/s0022-510x(98)00304-9.
[38] I. G. McKeith et al., “Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop.,” Neurology, vol. 47, no. 5, pp. 1113–1124, Nov. 1996, doi: 10.1212/wnl.47.5.1113.
[39] F. J. Jiménez-Jiménez et al., “Drug-induced parkinsonism in a movement disorders unit: A four-year survey.,” Parkinsonism Relat. Disord., vol. 2, no. 3, pp. 145–149, Jul. 1996, doi: 10.1016/1353-8020(96)00013-2.
[40] P. A. LeWitt and S. Fahn, “Levodopa therapy for Parkinson disease: A look backward and forward.,” Neurology, vol. 86, no. 14 Suppl 1, pp. S3-12, Apr. 2016, doi: 10.1212/WNL.0000000000002509.
[41] J. Jankovic and W. Poewe, “Therapies in Parkinson’s disease.,” Curr. Opin. Neurol., vol. 25, no. 4, pp. 433–447, Aug. 2012, doi: 10.1097/WCO.0b013e3283542fc2.
[42] J. P. Frankel, A. J. Lees, P. A. Kempster, and G. M. Stern, “Subcutaneous apomorphine in the treatment of Parkinson’s disease.,” J. Neurol. Neurosurg. Psychiatry, vol. 53, no. 2, pp. 96–101, Feb. 1990, doi: 10.1136/jnnp.53.2.96.
[43] T. Müller, “Catechol-O-methyltransferase inhibitors in Parkinson’s disease.,” Drugs, vol. 75, no. 2, pp. 157–174, Feb. 2015, doi: 10.1007/s40265-014-0343-0.
[44] L. V Kalia, J. M. Brotchie, and S. H. Fox, “Novel nondopaminergic targets for motor features of Parkinson’s disease: review of recent trials.,” Mov. Disord., vol. 28, no. 2, pp. 131–144, Feb. 2013, doi: 10.1002/mds.25273.
[45] S.-41. https://doi. org/10. 1002/mds. 2382. Fox, Susan HFox, S. H., Katzenschlager, R., Lim, S.-Y., Ravina, B., Seppi, K., Coelho, M., Poewe, W., Rascol, O., Goetz, C. G., & Sampaio, C. (2011). The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the motor symptoms of et al., “The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the motor symptoms of Parkinson’s disease.,” Mov. Disord., vol. 26 Suppl 3, pp. S2-41, Oct. 2011, doi: 10.1002/mds.23829.
[46] M. Emre et al., “Rivastigmine for dementia associated with Parkinson’s disease.,” N. Engl. J. Med., vol. 351, no. 24, pp. 2509–2518, Dec. 2004, doi: 10.1056/NEJMoa041470.
[47] S. Perez-Lloret, M. V. Rey, A. Pavy-Le Traon, and O. Rascol, “Emerging drugs for autonomic dysfunction in Parkinson’s disease.,” Expert Opin. Emerg. Drugs, vol. 18, no. 1, pp. 39–53, Mar. 2013, doi: 10.1517/14728214.2013.766168.
[48] W. M. McDonald, I. H. Richard, and M. R. DeLong, “Prevalence, etiology, and treatment of depression in Parkinson’s disease.,” Biol. Psychiatry, vol. 54, no. 3, pp. 363–375, Aug. 2003, doi: 10.1016/s0006-3223(03)00530-4.
[49] S. K. Kalia, T. Sankar, and A. M. Lozano, “Deep brain stimulation for Parkinson’s disease and other movement disorders.,” Curr. Opin. Neurol., vol. 26, no. 4, pp. 374–380, Aug. 2013, doi: 10.1097/WCO.0b013e3283632d08.
[50] A. L. Benabid, P. Pollak, E. Seigneuret, D. Hoffmann, E. Gay, and J. Perret, “Chronic VIM thalamic stimulation in Parkinson’s disease, essential tremor and extra-pyramidal dyskinesias.,” Acta Neurochir. Suppl. (Wien)., vol. 58, pp. 39–44, 1993, doi: 10.1007/978-3-7091-9297-9_8.
[51] Radiopaedia, “DBS.” https://radiopaedia.org/cases/deep-brain-stimulation-for-parkinson-disease?lang=gb.
[52] B. E. Fisher et al., “The effect of exercise training in improving motor performance and corticomotor excitability in people with early Parkinson’s disease.,” Arch. Phys. Med. Rehabil., vol. 89, no. 7, pp. 1221–1229, Jul. 2008, doi: 10.1016/j.apmr.2008.01.013.
[53] H. Chen et al., “Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease.,” Arch. Neurol., vol. 60, no. 8, pp. 1059–1064, Aug. 2003, doi: 10.1001/archneur.60.8.1059.
[54] M. C. de Rijk et al., “Dietary antioxidants and Parkinson disease. The Rotterdam Study.,” Arch. Neurol., vol. 54, no. 6, pp. 762–765, Jun. 1997, doi: 10.1001/archneur.1997.00550180070015.
[55] A. A. Farooqui and L. A. Horrocks, “Lipid peroxides in the free radical pathophysiology of brain diseases.,” Cell. Mol. Neurobiol., vol. 18, no. 6, pp. 599–608, Dec. 1998, doi: 10.1023/a:1020625717298.
[56] K. M. Powers, T. Smith-Weller, G. M. Franklin, W. T. J. Longstreth, P. D. Swanson, and H. Checkoway, “Parkinson’s disease risks associated with dietary iron, manganese, and other nutrient intakes.,” Neurology, vol. 60, no. 11, pp. 1761–1766, Jun. 2003, doi: 10.1212/01.wnl.0000068021.13945.7f.
[57] C. H. Williams-Gray et al., “The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort.,” J. Neurol. Neurosurg. Psychiatry, vol. 84, no. 11, pp. 1258–1264, Nov. 2013, doi: 10.1136/jnnp-2013-305277.
[58] K. B. Mark Edwards, Niall Quinn, “Parkinson’s Disease and Other Movement Disorders,” 2e ed., Oxford University Press, 2008, pp. 18–34.