Article topic: Chorea and its Most Common Associated Pathologies
Author: Rama Amourah
Editor: Haneen Al-Abdallat, Haneen A. Banihani
Reviewer: Ethar Hazaimeh
Keywords: Huntington’s disease, Neuroacanthocytosis, Acquired chorea, Sydenham’s chorea, Movement disorders
Overview
Chorea is a distinct and debilitating hyperkinetic movement disorder that can result from various diseases. As a result, the underlying pathophysiology, prognosis, and treatment options of people who present with chorea are highly variable and can only be determined by further investigation. This article will focus on the most common causes of chorea, which can be split into two categories: primary; in which chorea is a classic symptom of a hereditary disorder; and secondary, in which the chorea is due to an exogenous insult or a hereditary disease that does not typically present with chorea. Understanding the most common causes of chorea and how they differ from each other in history, presentation, and further testing allows for more efficient and effective diagnosis and treatment of patients who suffer from this symptom.
Pathophysiology
The pathophysiology of chorea can be understood to be caused by overexcitation or insufficient inhibition of the excitatory pathways in the basal ganglia. It is commonly caused by damage to the basal ganglia leading to decreased function of the indirect inhibitory pathway, resulting in overexcitation of the direct pathway and hyperkinesia ¹. Dysregulation of basal ganglia pathways can be mediated by various endogenous and exogenous factors, including drugs, antibodies, ischemia (such as in stroke), dysfunctional metabolism, and aggregation of proteins ² ³ ⁴.
Causes of chorea
Causes of chorea can be divided into primary and secondary.
Primary Chorea
- Huntington’s disease is an autosomal dominant hereditary neurodegenerative disorder caused by trinucleotide repeat expansions in the huntingtin gene on chromosome 4 (> 36 repeats). The resultant mutated huntingtin protein accumulates in the basal ganglia, leading to neurodegeneration which affects the putamen and head of the caudate nucleus (Figure 1)³ ⁵. A systematic review of 2,174 papers concerning neuroimaging of Huntington’s disease showed that the disease was associated with findings of decreased glucose metabolism, perfusion, and D1 and D2 receptor binding in the basal ganglia ². It is the most common primary chorea, with the incidence being at least 4 cases per 100,000 people in the United States, Australia, and most European countries, sometimes reaching as high as 7 to 10 per 100,000 people ⁶. Huntington’s disease is diagnosed through genetic testing, and clinical treatment is guided by the Total Functional Capacity (TFC) scale, which assesses a person’s ability to independently perform occupational, financial, domestic, and daily life tasks ⁷.
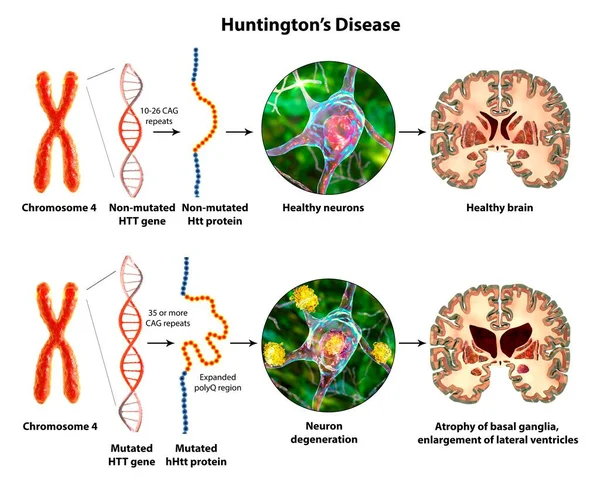
Figure (1): 3D illustration of Huntington’s disease
- Neuroacanthocytosis is an umbrella term for multisystemic hereditary disorders characterized by acanthocytosis (thorny appearance of red blood cells due to membrane protein defects) and degeneration of the basal ganglia, leading to various movement disorders, including chorea ⁸. Two well-studied diseases in this group which are most strongly associated with acanthocytosis and neurological manifestations are McLeod syndrome and chorea-acanthocytosis, which are commonly referred to as “core diseases” ⁹. McLeod syndrome is an X-linked recessive hereditary disorder caused by a mutation in the XK gene, which codes for a protein that may function in membrane transport associated with a lack of Kell antigens on the surface of red blood cells ¹⁰ ¹¹. Chorea-acanthocytosis (Figure 2) results from a hereditary autosomal-recessive mutation of the vacuolar protein sorting gene 13A gene (VPS13A) on chromosome 9q21, which leads to an absence of the ubiquitous VPS13A protein (also known as chorein). Chorein is still being studied, and little is understood about it apart from its role as a membrane transport protein ¹² ¹³.
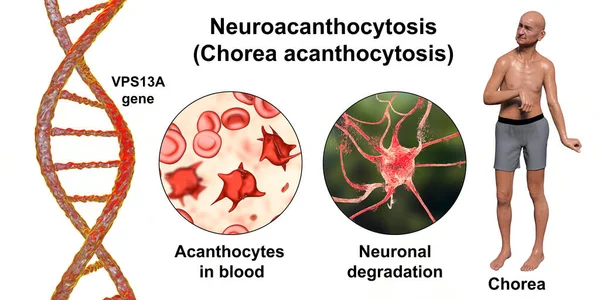
Figure (2): 3D illustration of Neuroacanthocytosis
Secondary Chorea
- Sydenham’s chorea of rheumatic fever affects 20% of children and adolescents with rheumatic fever, and disproportionately children in socioeconomic circumstances where treatment and prophylaxis are difficult to access ¹⁴. Rheumatic fever is caused by antibodies produced by the host immune system against group A beta-hemolytic streptococcus species (GAS). These antibodies persist even after the initial infection is cleared due to antigen cross-reactivity between the various M protein subtypes of GAS and different human tissues, resulting in the classical symptoms described by the Jones criteria, including Sydenham’s chorea ¹⁵ ¹⁶. It is most commonly seen in children aged 5 through 9 of both sexes and affects more post-pubertal females than males ¹⁷. Sydenham’s chorea is most likely caused by cross-reactivity between GAS M-protein antigens and antigens of the basal ganglia and its dopamine receptors, resulting in infiltration and neuronal loss ¹⁸. While Sydenham’s chorea is classically expected to self-resolve within 1 – 6 months, evidence shows that treating it results in higher rates of remission and reduced socioeconomic burden due to reduced ability to participate in school, work, and society ¹⁹ ²⁰.
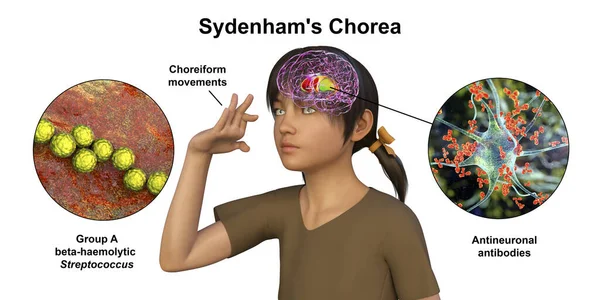
Figure (3): 3D illustration of Sydenham’s Chorea
- Chorea as a complication of autoimmune diseases; Systemic lupus erythematosus (SLE), antiphospholipid syndrome (APS), and celiac disease are three autoimmune diseases noted to have chorea as a complication. Up to 50% of SLE patients have neurological manifestations, but only 2% of those 50% develop chorea. Up to 25% of people with SLE have antiphospholipid antibodies, and that subset of SLE patients are more likely to develop antibody-induced chorea. The presence of antiphospholipid antibodies means that APS and SLE-associated chorea have similar pathophysiologies and complications. SLE and APS patients with chorea are at risk of arterial thrombosis secondary to a compromised blood-brain barrier, which can lead to cerebrovascular ischemia. There are a variety of proposed mechanisms, such as antibody-mediated platelet damage increasing platelet adhesion or antibodies preventing protein C from inhibiting thrombus formation. They are also both liable to worsen with oral contraceptives and pregnancy. People with celiac disease rarely experience chorea due to anti-gliadin antibodies crossing the blood-brain barrier and damaging the basal ganglia. In this case, the most effective treatment is ensuring the patient stops eating gluten ²¹ ²² ²³.
- Chorea as a complication of metabolic abnormalities; hereditary metabolic disorders may present with chorea, but it is rare. The hereditary metabolic disorder most notable for presenting with chorea is Wilson’s disease, an autosomal recessive defect on chromosome 13q14.3, which leads to defective ceruloplasmin and impaired ability to excrete copper. Chorea in Wilson’s disease and other hereditary metabolic disorders usually presents with other neurological symptoms and movement disorders. Many other metabolic abnormalities may present with chorea, a few of which are vitamin B12 deficiency, hyperthyroidism, hypoparathyroidism, hyper- or hypoglycemia, and hyper- or hyponatremia ⁴ ²⁴.
- Drug-induced chorea; Alcohol, central nervous system stimulants, dopamine antagonists, benzodiazepines, antiepileptics, oral contraceptives, and many other drugs may induce chorea, possibly by overstimulating direct pathway neurons and postsynaptic dopamine receptors or by causing hypersensitization of postsynaptic dopamine receptors ²⁵ ²⁶. Levodopa, the first-line treatment for Parkinson’s disease, is associated with various movement disorders such as tardive dyskinesia and has been found to induce chorea in 40% or more of patients taking it. Levodopa does not cause chorea in people who do not already have basal ganglia damage ²⁷. Chorea induced by oral contraceptives only appears where there is prior antibody-mediated basal ganglia damage and, as such, is associated with a history of Sydenham’s chorea, chorea of SLE, and chorea gravidarum ²⁵ ²⁸.
Presentation
Chorea typically presents as involuntary, flowing, “dance-like” movements. (Figure 4). In comparison to some other hyperkinetic like Ballism (hemiballism if unilateral) is characterized by more proximal and extreme movements, often described in the literature as “flinging” in nature. Hemiballism and ballism are associated with decreased subthalamic function or hyperactivity of the globus pallidus internus ²⁶ ²⁹. Athetosis, which may also be referred to as dystonia with some choreic quality, is often characterized in the literature as a more slow and writhing motion than chorea. Cerebral palsy, kernicterus (bilirubin encephalopathy), stroke, neonatal asphyxia, and trauma are commonly associated with athetosis ²⁶ ³⁰.
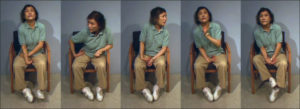
Figure (4): chorea movement
Differential diagnoses
Huntington’s disease is most likely to appear in patients between 35 and 40 years old and is characterized by a suggestive family history, cognitive decline, personality changes, dementia, and many other psychiatric disturbances such as depression and suicidality ³¹. Cognitive decline and psychiatric symptoms are the first signs to appear. Hyperreflexia, orofacial dystonia (such as involuntary tongue movements), and irregular eye movement precede chorea, with rigidity and parkinsonism typically presenting late in the course of the disease. Orofacial dystonia and abnormal, poorly controlled eye movement are also common findings. Patients who present at twenty years old or younger (juvenile Huntington’s) are more likely to present with bradykinesia and parkinsonism than chorea, and they usually have an affected father ⁴ ⁵ ³² ³³.
Core neuroacanthocytosis (McLeod syndrome and chorea-acanthocytosis) present similarly to Huntington’s disease as that cognitive and psychiatric symptoms precede chorea, hyperkinesia, and parkinsonism happens late in the course of the disease. However, they are both characterized by dystonia, peripheral neuropathy, hypo- or areflexia, seizures, and acanthocytes on a peripheral blood smear (Table 2) ³⁴. Cognitive decline is markedly reduced compared to that seen in Huntington’s disease ⁴. McLeod syndrome, in particular, is characterized by elevated creatinine kinase due to myopathy and elevated lactate dehydrogenase and may have cardiac manifestations such as dilated cardiomyopathy ³⁵. Patients with McLeod syndrome may experience a hemolytic transfusion reaction if the blood donor expresses Kell antigens normally ³⁶. Choreic neck and truncal flexion, which presents as “head drop,” as well as orofacial involvement characterized by tongue biting, dysphagia, and dysarthria, are characteristic of chorea-acanthocytosis ⁹ ³⁷.
Table (1) ⁸ ⁹. Characteristics of McLeod syndrome and chorea-acanthocytosis, two core neuroacanthocytosis syndromes.
| McLeod syndrome | Chorea-acanthocytosis | |
| Gene
Protein Inheritance Onset Acanthocytes Serum creatinine kinase (IU/L) Chorea Other motor signs Seizures Cardiac symptoms Neuromuscular signs Unique transfusion reactions |
XK
XK X-linked recessive Usually ages 20 – 30 Strong association 300 – 3,000 Strong association Involuntary vocalizations Generalized Dilated cardiomyopathy, arrhythmias Areflexia, weakness, atrophy Kell positive donors |
VPS13A
Chorein Autosomal recessive Usually ages 25 – 60 Strong association 300 – 3,000 Strong association Orofacial dystonia, bradykinesia, and rigidity, gait dystonia Partial-complex, generalized None Areflexia, weakness, atrophy None |
Sydenham’s chorea should be considered when a child with chorea between five and fifteen years of age presents with a history of recent infection, symptoms, and signs within the Jones criteria for rheumatic fever (Table 2), or a high antistreptolysin O titer indicating recent group A beta-hemolytic streptococcal infection ³⁸.
Drug-induced chorea should be considered based on drug history, alcohol consumption, and a previous history of chorea or basal ganglia damage ²⁶.
Table 2 ³⁹. Modified 1992 Jones criteria for diagnosing rheumatic fever.
| Major (diagnostic: 2, 1 and 2 minor) | Minor |
| Carditis
Arthritis Sydenham’s chorea Subcutaneous nodules Erythema marginatum |
Arthralgia
Hyperpyrexia Elevated CRP and ESR Prolonged P-R interval |
Chorea due to autoimmune diseases such as antiphospholipid syndrome (APS), celiac disease, and systemic lupus erythematosus (SLE) may present with a medical history or other symptoms which are characteristic of autoimmune diseases such as rashes, arthralgia, thrombosis in cases of SLE and APS, and digestive symptoms in celiac disease. Testing for antibodies and other laboratory findings of autoimmune disease is also helpful ²¹.
Wilson’s disease is a differential diagnosis in patients under 40 who present with chorea. A significant finding of Wilson’s disease is Kayser-Fleischer rings, which are brown rings surrounding the cornea due to copper deposits (Figure 5). Low serum ceruloplasmin, high copper excretion, and hepatic injury due to copper deposits are also suggestive. Blood and genetic testing can confirm a diagnosis of Wilson’s disease ⁴. Other metabolic abnormalities should also be considered in children with chorea who do not have a history or findings suggestive of Sydenham’s chorea. They may be confirmed by laboratory investigation, past medical and family history, characteristic associated symptoms, and genetic testing in cases of hereditary metabolic disorders.
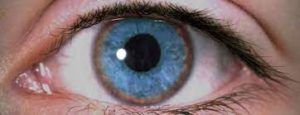
Figure (5): Kayser-Fleischer ring
Drug and social history should be considered in case of drug-induced chorea without other risk factors.
Management
It is essential to address the underlying cause of secondary chorea, while primary chorea can only be treated symptomatically.
Primary
The underlying genetic diseases associated with primary chorea are best addressed in a multidisciplinary manner, in addition to the symptomatic treatment of chorea mentioned above. Huntington’s disease, for example, may be addressed through patient education, assistive devices and clothing, and various therapies, including occupational therapy, physiotherapy, psychological therapy, and speech therapy ³¹. This also applies to neuroacanthocytosis diseases such as McLeod syndrome and chorea-acanthocytosis, such as speech therapy and specialized feeding for people with orofacial dystonia associated with chorea-acanthocytosis. In McLeod syndrome, special care must be taken to prevent transfusion reactions and cardiologic complications. Dopamine depleters and antagonists such as tetrabenazine are effective in managing chorea associated with both Huntington’s disease and neuroacanthocytosis; however, care must be taken in patients with Huntington’s disease for increased suicidality. Antiepileptics and neuroleptics are used to ameliorate chorea, treat neuroacanthocytosis seizures, and ameliorate Huntington’s disease’s psychiatric complications ¹ ⁹ ³⁵ ²⁵ ⁴⁰.
Secondary
As diverse as the causes of secondary chorea are, they all have in common that addressing the underlying insult is key to management, and most secondary chorea stop once the initial cause is treated.
The first and most important step in managing Sydenham’s chorea is to address the underlying streptococcal infection. Intramuscular penicillin G is the first line of treatment, and macrolides such as erythromycin are used for patients allergic to penicillin ⁴¹ ²⁰. Several drugs have been used to treat Sydenham’s chorea, including dopamine antagonists such as haloperidol, antiepileptics such as valproic acid, and steroids ⁴² ⁴³. While several institutions classically recommend dopamine antagonists, antiepileptics are favored by more recent sources for having fewer associated adverse effects and a similar response rate ¹⁴ ⁴¹ ¹.
Chorea secondary to systemic lupus erythematosus and the primary antiphospholipid syndrome is best treated with valproic acid, tetrabenazine, haloperidol, and corticosteroids. Aspirin is also helpful in preventing thrombosis and its complications. Chorea due to celiac disease usually stops when the patient stops eating gluten and receives treatment ²¹ ²³.
Wilson’s disease is treatable with copper-chelating agents such as penicillamine. Metabolic chorea usually stops once the underlying disorder or deficiency is treated ⁴. Most drug-induced chorea stop with the withdrawal of the drug; however, movement disorders may persist after drug withdrawal.
Prevention
Primary
Given that the primary choreas mentioned are hereditary, patient education and genetic counseling are essential to give patients the ability to make informed reproductive choices. In addition, prenatal testing is also an option for patients to anticipate their children’s future medical needs ³¹ ⁹.
Secondary
Prevention of secondary chorea varies based on the nature of the primary insult. Sydenham’s chorea can be prevented by antibiotic prophylaxis of penicillin G, which protects against GAS infections and rheumatic fever ²⁰. Diagnosing and treating metabolic and autoimmune disorders minimizes, if not resolves, most of their associated symptoms, including more severe sequelae such as chorea.
Risk Factors
Primary
Family history is the leading risk factor for the primary chorea listed, as they are hereditary.
Secondary
Autoimmune diseases such as rheumatic fever secondary to GAS infection, SLE, and celiac disease are all risk factors for autoimmune-mediated chorea. Basal ganglia pathologies, including previous cases of chorea, will increase the risk of chorea in patients taking levodopa and oral contraceptives. Hereditary metabolic disorders and acquired deficiencies and abnormalities pose a small but significant risk of patients developing chorea. People taking central nervous stimulants, dopamine antagonists, antiepileptics, benzodiazepines, and other drugs are at risk of developing chorea even without a history of chorea or basal ganglia pathology ⁴ ²⁵ ²⁴.
Prognosis
Primary chorea persists for life and cannot be cured. For example, in Huntington’s disease, neurodegeneration progresses until it stabilizes after 15 – 30 years or the patient dies. Patients with Huntington’s disease usually survive 10 to 30 years after diagnosis, and their most common cause of death is aspiration pneumonia ³¹.
Neuroacanthocytosis syndromes vary widely in prognosis. McLeod syndrome has a mean onset of around 30 years old, and patient life expectancy depends on the presence, severity, and management of neurological and cardiac sequelae ⁴⁴. Chorea-acanthocytosis has an average onset of 35 years old, although there is more variation in onset age and severity than there is for McLeod syndrome ⁸ ⁹.
Most secondary chorea resolves with no further problems once the underlying insult or disease is addressed. The patient’s prognosis depends not on the chorea but on the pathology that caused it.
Recent Developments
Deep brain stimulation (DBS) of various basal nuclei, particularly the globus pallidus internus, has been demonstrated to improve chorea in most Huntington’s and non-Huntington’s disease neurodegenerative disorders patients, such as chorea-acanthocytosis ⁴⁵ ⁴⁶ ³⁷. However, the evidence of the long-term efficacy of DBS is scarce, and there are concerns of high publication bias in favor of that evidence ³².
Chorein (VPS13A protein) and XK protein were recently found to form a complex and be involved in the same molecular pathways. The function of this lipid transport complex is unclear, but it is likely essential to the structure and function of both neurons and red blood cells. These findings indicate that McLeod syndrome and chorea-acanthocytosis are more closely linked than previously thought and may shape future understanding of neuroacanthocytosis syndromes and their pathophysiologies ¹³.