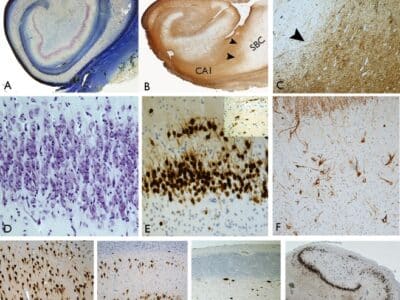
Article topic: Lambert Eaton myasthenic syndrome (LEMS)
Authors: Lobana Nabeel Mahdawi, Samah Zeidan Alajjawe
Editor: Lubna AL-Rawabdeh
Reviewer: Ethar Hazaimeh
keywords: Neuromuscular junction, Lung Cancer, Autoantibodies, LEMS.
Overview
Lambert Eaton myasthenic syndrome (LEMS) is considered a rare neuromuscular junction disorder that could be an idiopathic autoimmune disorder or a paraneoplastic phenomenon [1]. It is a rare disease with a well-characterized pathogenesis [2]. autoantibodies against presynaptic voltage-gated calcium channels that are present in the neuromuscular junction are the primary underlying mechanisms [3]. More than half of cases present as a paraneoplastic form associated with a malignant tumor which is a small cell lung carcinoma (SCLC) in most of the patients. The remaining cases are autoimmune disorders and frequently overlap with other autoimmune diseases [4]. Increased awareness and knowledge of the disease have improved the diagnostic methods decreased misdiagnoses and enhanced effective treatment of the disease or an early starting of oncological treatment that is associated with the disease [4].
Etiology
LEMS is caused by pathogenic autoantibodies to presynaptic voltage-gated calcium channels (VGCCs) in the membrane of the motor nerve terminal, preventing acetylcholine release, and leading to distinct weakness of striated skeletal muscles [2].
Lambert Eaton myasthenic syndrome is divided into paraneoplastic and non-paraneoplastic, also known as a nontumor Lambert Eaton myasthenic syndrome that is not associated with malignancy. Sixty percent of patients who develop Lambert-Eaton myasthenic syndrome have an underlying malignancy. Paraneoplastic type of Lambert-Eaton myasthenic syndrome primarily happens in association with small cell lung cancer (SCLC) [2]. Although there have been a few reports of non-small-cell and mixed-lung carcinomas. A lot of papers describe associations of LEMS with non-lung-cancer tumors. (e.g., prostate carcinoma, thymoma, and lymphoproliferative disorders) [4]. A history of smoking is also considered a risk factor for the disease. About 65% of young patients with NT–L EMS were found to have a genetic HLA-B8-DR3 haplotype [1]
Epidemiology
Lambert-Eaton myasthenic syndrome is known as a rare disorder of neuromuscular junction, with a prevalence of 46 times less than Myasthenia Gravis. However, the incidence of Lambert-Eaton myasthenic syndrome was 10 to 14 times less than the incidence of myasthenia gravies. increasing in the prevalence of myasthenia graves compared to Lambert-Eaton myasthenic syndrome indicates the poor prognosis and survival of Lambert-Eaton myasthenic syndrome specifically when associated with SCLC [1]. A low prevalence relative to incidence indicates the poor survival of LEMS patients with the paraneoplastic type of disease. Male represents 60% of the LEMS patients. The mean age of diagnosis was 58 years. There are two peaks for the age of disease onset, one around 40 years and one at a higher age around 60, similar to what is seen for myasthenia gravis disease [2]. The Lambert-Eaton myasthenic syndrome not related to malignancy has a nearly normal survival rate [1].
Pathophysiology
Lambert-Eaton myasthenic syndrome is a disorder of decreased acetylcholine release from the presynaptic nerve terminals due to antibodies against voltage-gated calcium channels in the presynaptic neuronal cell membrane [1]. Such antibodies show a high sensitivity, as they can be detected in 85% of all LEMS patients. The LEMS in patients who have distinct muscle weakness is close to 100%. Among SCLC patients without any symptoms of muscle weakness or autonomic dysfunction, 3–5% have VGCC antibodies [2].
Antibodies to P/Q-type VGCC are very specific to LEMS but have been found in 1–4% of patients with small cell lung cancer without neurological problems. Similarly, these antibodies are also detected in the serum and CSF of a few patients with subacute cerebellar ataxia, both with and without clinical symptoms of LEMS, nearly all of whom had an associated small cell lung cancer [4]. Rarely, antibodies for the N-type VGCC have been detected in malignancy-associated Lambert-Eaton myasthenic syndrome [1].
Antibodies against SOX-1 were also used as a serological tumor marker with diagnostic value to differentiate between Paraneoplastic LEMS and Autoimmune LEMS. These antibodies have been detected in 64% of patients with Para neoplastic LEMS and only 0–5% of Autoimmune LEMS patients according to some studies and are therefore able to identify LEMS patients with a greater risk of malignancy [5].
Genetic factors: Non-tumor Lambert-Eaton myasthenic syndrome is associated with HLA-B8 and HLA -DR3 and -DQ2 These HLA genotypes have also been found in other autoimmune conditions including myasthenia gravis. However, it is not found in Lambert-Eaton myasthenic syndrome that happens with SCLC [1].
Clinical presentation
The presentation of LEMS is progressive and varies significantly, but patients usually present with proximal muscle weakness, and autonomic disturbances such as dry mouth, constipation, diplopia and dysarthria, and areflexia. Additionally, some patients address some respiratory and sensory symptoms [6][7].
Symptomatic autonomic dysfunction has been found in most patients and precedes the onset of muscle weakness for years. There are many symptoms that patients come with, but the most common are dry mouth, constipation, and erectile dysfunction in men [7][4]. Other less common symptoms: are dry eyes, orthostatic hypotension, and urinary dysfunction so daily activities are affected significantly [10]. Many muscles can be affected by LEMS, upper legs and hips muscles are the most involved muscles, and the weakness of muscles starts proximally and then spreads distally to include hands and feet muscles and caudally to cranially [8][4].
Diagnosis
The diagnosis is established depending on the combined clinical presentation, electrophysiological study, and immunological study that will reveal the VGCC antibodies [7][8].
The presence of the typical triad of proximal muscle weakness, areflexia, and features of the dysfunctional autonomic system, make the first step in terms of diagnosis that must be confirmed by electrophysiological and immunological studies [9].
Among the electrophysiological studies, Repetitive nerve stimulation (RNS) is the most significant to diagnose LEMS, in which it will reveal the reduced amplitude of the first compound muscle action potential (CMAP) in LEMS patients, decreased the response of low-frequency repetitive nerve stimulation, and increased the response following short exercise [4][11]. However, there are many other electrophysiological findings have been observed [6].
Since LEMS clinical manifestations result from the action of autoantibodies against VGCC on the presynaptic nerve terminals preventing the release of acetylcholine, the detection of these antibodies strongly supports the diagnosis of LEMS. Nearly,80-90% of patients have high serum titer, making the autoantibodies a reliable diagnostic marker [6][4].
Since MG is on the top of the differential diagnosis of LEMS, MG usually starts with ptosis and diplopia, while LEMS often presents mild upper leg weakness. Moreover, the muscle weakness in MG spreads in a craniocaudal pattern and autonomic changes and areflexia are much less common than in patients with LEMS [4]. The diagnosis of the LEMS can be challenging since its clinical pattern is less specific and difficult to recognize [4].
Management
In terms of treatment, LEMS isn’t a curable disease and there are only a few options of medication for symptomatic relief. The most common one is 3,4-diamino pyridine which inhibits the voltage-gated k+ channel to extend the presynaptic action potential to produce more VGCC to open. Since it can be used alone or with an acetylcholinesterase inhibitor, patients won’t return to their normal activity ultimately [8].
When patients fail to improve on 3,4-DAP, the immunomodulatory strategies come on the second rank, and include, immunosuppressive drugs, intravenous immunoglobulin (IVIg), and plasma exchange [8].
References
References...