Article Topic: Myasthenia gravis
Author name: Basel Abbas Asad Ghwini
Keyword: autoimmune – acetylcholine receptors – antibodies – myasthenia gravis
Overview and epidemiology
Thomas Willis wrote about a woman who lost her ability to speak in his book “De anima brutorum” , the book was published in 16721, he was the first to write a description about myasthenia gravis (MG), on the other hand Wilks was the first person who report myasthenia gravis in 18772. At the end of 19th century, erb and goldflam described the myasthenia gravis clinical symptoms 3.it was named as erb-goldflam syndrome for several years. In 1895, Jolly used the name of myasthenia gravis and explained myasthenic reaction of muscle 3.
Myasthenia gravis is an autoimmune chronic disease in which antibodies are directed against acetylcholine receptors or against proteins involved in the clustering of acetylcholine receptors (MuSK, LRP-4, or agrin) in the postsynaptic membrane at the neuromuscular junction and leads to skeletal muscle weakness and fatigue4,5.
The prevalence is about 140 cases per million6,7, with an annual incidence estimated to be between ten and thirty per million6,8. the peak of incidence is different between men and women, with a bimodal distribution, women is more commonly affected under the age of forty, and men more commonly affected over the age of fifty6,9. The frequency of myasthenia gravis in the elderly seems to be rising recently, attributed to enhanced awareness of the disease in recent decades6.
Pathogenesis and etiology
When action potential travel through alpha motor neurons and reaches the presynaptic boutons, which contains acetylcholine loaded synaptic vesicles10, depolarization of presynaptic membrane opens voltage gated calcium channels, causing the calcium ions to influx, calcium then docking of the synaptic vesicles to the presynaptic terminals, enabling release of Ach into the synaptic cleft, binding of acetylcholine to acetylcholine receptors on the muscle membrane leads to sodium and potassium flux across the junctional membrane, leading to depolarization of endplate. If the motor endplate potential (EPP) reaches threshold, voltage gated sodium channels located at the underside of the folds are opened, resulting in sodium influx and therefore the generation of an “all or none” muscle fiber action potential that spreads bidirectionally through the muscle fiber. In myasthenia gravis patients, A reduction of AChR molecules causing “reduced safety factor” , and the number of AChRs contacted may be inadequate to drive the EPP to threshold, causing failure of neuromuscular transmission6,11.
The disease is divided into several sub-groups because the clinical manifestations of myasthenia gravis vary according to the age of onset, the antibody involved and the presence of thymic pathology 12.
A. Pure ocular form
Around 15% of myasthenia gravis patients present with only ocular symptoms12, due to high percentage of patients with initial ocular manifestation in the first year of onset, minimally 2 years delay without generalization is needed to classify a patient as a pure ocular form. 50% of these patients present antibodies not detectable by a classical assay but often demonstrable in a cell-based assay “CBA”13.
B. Generalized form with anti AChR antibodies.
Around 85% of myasthenia gravis patient displays this form12. There is no apparent correlation between the level of antibodies and the severity of the disease. Thymic abnormalities are usually found in these patients, and the maximum titers of antibodies are seen in patients with thymic follicular hyperplasia, the anti-AChR antibodies belong to the IgG1 and IgG3 subclasses14. The patients with AChR antibodies can be divided into two sub-groups:
- Early onset myasthenia gravis “EOMG”, the onset before the age of 50 and it is predominantly to women, mainly thymic follicular hyperplasia found in women15.
- Late onset myasthenia gravis “LOMG”, the onset after the age of 50, most patients generalized and severe symptoms like bulbar involvement 16.
C. The forms without classical anti-AChR antibodies
- The form with anti-MuSK antibodies
Around 5% of patients with myasthenia gravis has this antibody kind17, the patients are typically female, and they have serious form of the disease with common muscular atrophy, the respiratory muscles are commonly affected, but ocular symptoms and thymic abnormalities are unusual. The anti-MuSK antibodies belong to the IgG4 subclass18.
- The form with anti-LRP4 antibodies
Around 12-50%of the seronegative population presents antibodies to LRP4. These antibodies were detected in a subgroup of patients who were double seronegative for AChR and MuSK19.
- The form with clustered AChR antibodies
Around 5% of the myasthenia gravis patients has no recognized antibodies12. The antibodies can be discovered by cell-based assay, in which AChRs are assembled, the clinical pattern is similar to myasthenia gravis with classical anti-AChR antibodies involving ocular and generalized form⁏ thymic hyperplasia may possibly be present13.
D. Neonatal myasthenia gravis
There is a possibility of aggravation of the myasthenic symptoms in 20-40% of females throughout the first 3 months. In 10-20% of the cases, the newborn will display a transient neonatal myasthenia (TNM) that could last a few days to 3 months. This disease is because of the passive transfer of the antibodies from the mother, mainly the anti-AChR antibodies20.
Clinical presentation
Clinically, patients with myasthenia gravis will have nonspecific features of weakness and fatigability which are fluctuating in nature, these symptoms will get worse as the day goes on or with exercise, indicative of the loss of function of AChRs, and improves with rest21.
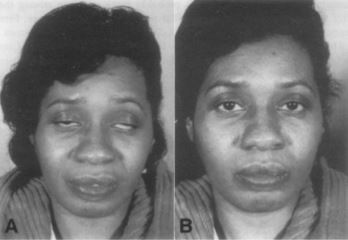
Patients with myasthenia gravis will also have a different pattern of weakness due to the vulnerability of certain muscle groups. Most of the patients will initially present with ocular symptoms like ptosis and diplopia but with no pupillary abnormalities 21(figure1). About 50% of patients will have ocular symptoms, and that is why most patients are initially seen by an ophthalmologist. Around 90% of patients eventually have ocular manifestation within 1 to 2 years22. Bulbar muscle weakness shows dysarthria, dysphagia, or chewing difficulties. Limp (appendicular)muscle weakness caused by myasthenia gravis is rare.
Exacerbation of symptoms appears with infection, pregnancy, menses, heat exposure. exacerbation that needs mechanical respiratory support is called myasthenia crisis 22. the occurrence of some severe symptoms like shortness of breath, ineffective cough, choking, and rapid motor deterioration indicate MG crisis which is an emergency and needs immediate hospitalization in intensive care.12
On neurological examination there may be positive findings of gaze palsy, which can be unilateral, and bilateral ptosis which will reduce facial expression and affect smile, lower lip may be slightly everted, jaw may drop, dysarthria and dysphonia can be present, hyperacusis due to weakness of inner ear stapedius muscle 22, Chewing difficulties may present because of jaw fatigue ,also swallowing may be affected due to pharyngeal muscle weakness 21 . limp weakness is usually proximal and asymmetrical, the flexor muscles of neck weaken early 22.
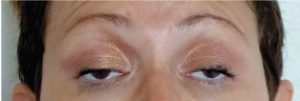
Figure 1: Bilateral asymmetrical ptosis in MG patients (24).

Diagnosis
To diagnose a patient with myasthenia gravis may be somehow challenging because of fluctuation character and as the fatigue is a common symptom of various neuromuscular disorders23.
Serologic testing is the first diagnostic step for MG. anti-acetylcholine receptor antibodies assay, purified AChR from obtained skeletal muscle is labeled with radio iodine alpha bungarotoxin23. Its sensitivity to generalized form of myasthenia gravis is about 85 percent, and to ocular form is about 50 percent. For patient with similar grade of weakness the serum concentration of acetylcholine antibodies differs broadly among myasthenia gravis patients. To diagnose myasthenia gravis clinical features are essential along with elevated concentration of antibodies in patient serum, although other diseases may have elevated serum AChR antibodies like autoimmune liver disease, systemic lupus, and in patients with thymoma, as well as in RA patients whom receiving penicillamine25.there are many other assays somehow increase the sensitivity like AChR-blocking antibodies which measures the capability of patient serum to inhibit cholinergic ligands binding , or AChR-modulating antibodies which measures the capability of patient serum to induce modulation of AChR in cell cultures26.
The low affinity anti -acetylcholine receptor antibodies in which antibodies binds to AChRs clustered on the surface of cells other than muscle is shown in about 66 percent generalized form MG patients who were negative for antibodies on all standard assays [AChR, MuSK]13.
Anti-striated muscle antibodies are highly related with thymoma, around 75 to 80 percent of myasthenia gravis patients along with thymoma are positive, in some cases it can be positive in non-thymomatous patients like in old patients27. There might be a correlation between the presence of anti-titin or anti-ryanodine antibodies and late-onset form of myasthenia gravis specifically with more severe disease28.
One of noninvasive and cost effective tools in diagnosing myasthenia gravis is ice pack test, which involves the application of a packet of ice onto the eyes for about 5 minutes, if ptosis resolves, the test is considered positive.29
One of short acting acetylcholine inhibitor which is Edrophonium chloride works by extending the duration of action of acetylcholine within the NMJ, it is used for patients suspected to possess myasthenia gravis. The edrophonium test, includes intravenous administration of edrophonium and then observation for progression in muscle strength (figure2), the edrophonium test sensitivity in MG diagnosis varies between 71% to 95% for generalized form30.
Figure 2: edrophonium test A bilateral ptosis in MG patient B IV edrophonium administration
Electrophysiological test can determine the exhaustion of neuromuscular transmission that caused by a diminished number of functional AChRs. Most frequently utilized is repetitive nerve stimulation in which repeated supramaximal electrical stimulation of a nerve is performed with recording of the compound muscle action potential in a subsequent muscle. There is 80% reduction in the amplitude or the area of the compound muscle action potential following repetitive nerve stimulation in patients with generalized form of myasthenia gravis , but less than fifty percent in patients with ocular form myasthenia gravis31.
Single-fiber electromyography [SFEMG] is a highly specialized manner that allows for recording of electrical potentials from single muscle fiber31. It can be considered if clinical suspicion for myasthenia gravis is high and all other testing were negative32,SFEMG can determine neuromuscular jitter, which is variation in the time electrical potential takes to reach the threshold for muscle fiber, in myasthenia gravis patients SFEMG shows abnormal jitter in 95-99 percent of patients if appropriate muscle are tested31, but SFEMG is not specific only to myasthenia gravis, other abnormalities in nerve or even in muscle can be seen23.
Chest CT scan is helpful in presence of thymoma, a generalize myasthenia gravis patient with anti-AChR antibodies needs to monitor the thymus every 5 year if the patient is not thymectomized12.
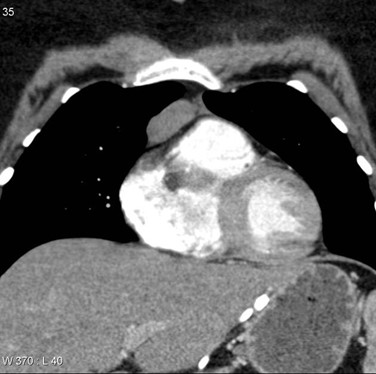
Figure 3: CT scan of the mediastinum of a young woman clinically demonstrate features of MG with anterior mediastinal thymoma type 2B, Case courtesy of Assoc Prof Frank Gaillard (33).
Differential diagnosis of MG
Include other neuromuscular junction disorders 34:
- lambert Eaton myasthenic syndrome
- Botulism
- Congenital myasthenic syndrome
- Acute inflammatory demyelinating polyradiculoneuropathy (AIDP) may encourage myasthenia gravis.
- Mitochondrial neuromuscular disorders
- Motor neuron disease
- Brainstem ischemia
- Organophosphate poisoning
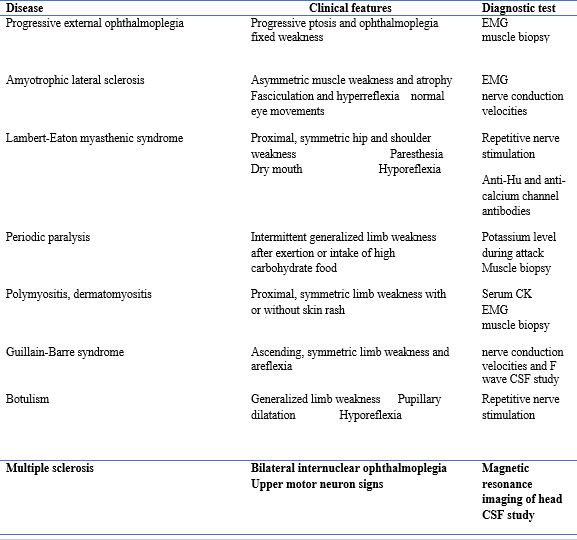
Table 2: differentiation of myasthenia and other neurological disorders22
Treatment
Treatment of myasthenia gravis is individualized, the overall goal of treatment is to restore the normal function and to minimize adverse effect of the disease, treatment selection depends on many factors including the distribution, duration, and the severity of the disease34. There might be complications associated with treatment, the risk of having these complication is increased with age, gender, medical comorbidities, and compliance of the patient34.
Plasma exchange and IVIG
Plasma exchange is used to decrease the number of circulating antibodies, and shows improvement in myasthenia gravis patient specifically the acquired form35.it is used as a short term treatment for severe cases, myasthenia gravis crisis, or prior thymectomy or another surgical procedures and in refractory cases of myasthenia gravis34. PE series consist of 5 to 6 exchanges of 2 or 3 liters on different days34. Many side effects could happened from PE includes paresthesia as a result of hypocalcemia and hypotension36.
The use of IVIg improve the muscle strength specially in severe cases, patient with septicemia and prior to surgeries, common side effect may include chills, headache, and fever. Some serious side effect may include renal toxicity, volume overload, aseptic meningitis and stroke but they are rare, Decreasing the rate of infusion may improve some side effects23. IVIGs is administered as 10% solution, with standard dosage is 2 gm/kg over 2 to 5 days34.practice parameter analyzed all controlled studies of thymectomy in myasthenia gravis patients, and the results were baffled by baseline variances between surgical and nonsurgical groups37. In a random trial compare between efficacy IVIg an PE, it support the use of IVIg in treatment of myasthenia gravis patients38.
Drugs for Symptomatic Therapy
There are several strategies to treat myasthenia gravis patients which includes improving the effect of acetylcholine by using cholinesterase inhibitors, get rid of AChR antibodies by plasma exchange or intravenous IG, thymectomy, immunosuppression23.
first-line medication used to treat myasthenia gravis patients is a drug that inhibits acetylcholine esterase function, due to its safety, rapid onset, few side effects. Pyridostigmine is the most used AChEI, it works as a symptomatic therapy by increasing the amount of ACh in the synaptic cleft of NMJ, starting dose of 30 to 60 every 4-6 hours34.
doses beyond 120 mg every 4 hours are not often effective and more likely to give cholinergic side effects include diarrhea, sweating, bradycardia, stomach cramps, increased secretions39,40.
Immunosuppressive Drug Therapy
Corticosteroids are commonly used in myasthenia gravis, prednisone is used as a first choice in treating myasthenia gravis, indicated to generalized form or ocular form myasthenia gravis patients that cholinesterase inhibitors were not enough to control their symptoms23,admenstration of prednisone is mostly at high doses 0.75-1.0 mg/kg/day for quite a few months at first, then gradual tapering off the dose. The improvement needs about 2-4 weeks to be observed23. Around 30-50% of patients showed transient worsening of symptoms when given high daily prednisone dose 41.corticosteroids have side effects in every system of the body like DM, osteoporosis, and obesity42. for most of the patients, azathioprine is added to corticosteroids, this combination will reduce the side effects and enhance the results, in case of contraindication for corticosteroids, azathioprine can be given as a monotherapy. The long term treatment is safe an nearly all patients43.
Most guidelines recommend mycophenolate mofetil for mild or moderate myasthenia gravis, the mechanism of action of this drug is by inhibiting purine synthesis and interference with B-cell and T-cell proliferation. Methotrexate, cyclosporine, and tacrolimus are alternative secondary immunosuppressive drugs43.
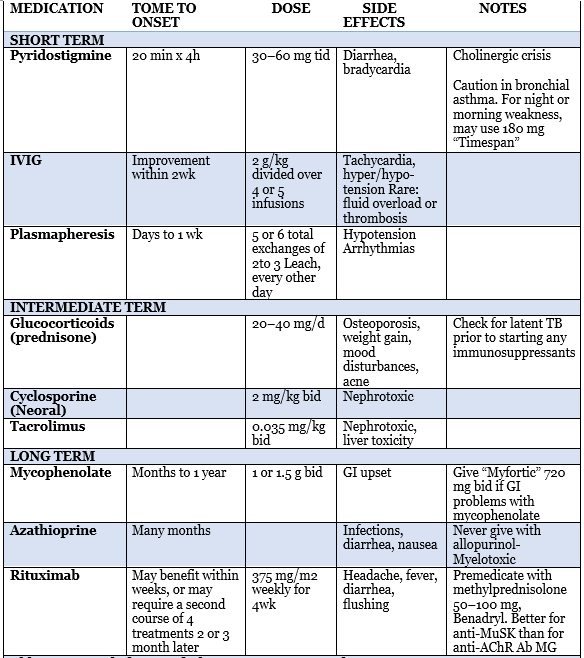
Abbreviations: bid, twice daily; GI, gastrointestinal; IVIG, intravenous immunoglobulin; TB, tuberculosis; tid, three times daily.
Table 3: myasthenia gravis treatment plan5
Thymectomy
Thymectomy is indicated for removal of a thymic tumor or as a treatment for MG. Thymic tumors can spread locally so they must be removed. Thymomas in general respond to corticosteroids treatment as well, for generalized myasthenia gravis thymectomy is recommended as a treatment, previous studies show that 1/3 of patients had excellent results, 1/3 improve, and 1/3 obtain no clinical benefit. Thymectomy should never be done as an emergent procedure, the earlier it performed, the more rapidly its benefits realized5.
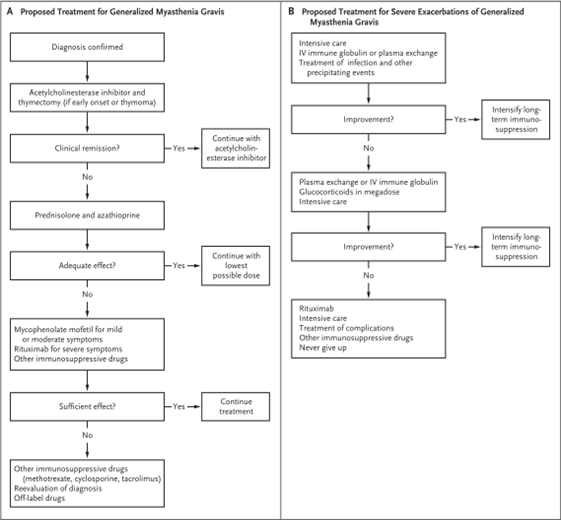
Figure 4: Algorithms for generalized MG treatment and for severe exacerbation for generalized disease43.
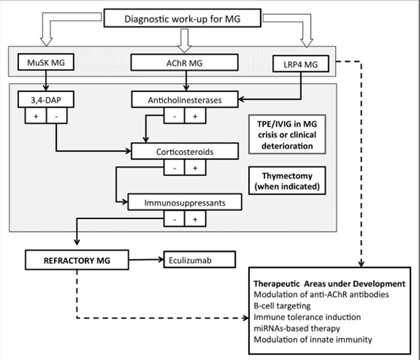
Figure 5: Flowchart summary of treatment strategies for the main serological subtypes of MG5
Refractory Myasthenia gravis
Even though most of myasthenia gravis patients respond to treatment listed above, some patients fail to respond to these agents, or cannot tolerate their side effect, treatment for those patients should try to remove the autoimmune response, by giving them high doses of cyclophosphamide to reboot their immune system. Many of them shows good results. This method necessitate special attention and should be performed under supervision of experts in the area of hematology/oncology5. Other choice can be given to those patients is Eculizumab, which is recombinant humanized monoclonal antibody that binds to the C5 complement protein and prevent the formation of C5b-9 membrane attack complex, candidates for this therapy are those with severe or refractory disease despite receiving adequate immunotherapies or can not tolerate their side effects, meningococcal vaccine should be given prior therapy administration44.
Myasthenic crisis
It is life-threatening manifestation of myasthenia gravis, weakness develop within minutes to days and involve flaccid tetra paresis, severe dyspnea, respiratory failure, and aspiration. Rapid progressive weakness of respiratory muscles is the main symptom. The early diagnosis and treatment the better outcome, treatment includes early intubation, it is essential to secure the airway, pyridostigmine or neostigmine to relief symptoms, plasmapheresis or immunoglobulins. Corticosteroids and azathioprine should be optimized. rituximab can be an option for escalation45.
Prognosis
Early in the coarse of myasthenia gravis, symptoms fluctuate and sometimes remit, even though such remission are rarely permanent, there are three major stages, an active stage described as relapses and remissions last about 7 years is followed by an inactive stage last about 10 years, the inactive stage is characterized by less disease volatility, however patients may have aggravation related to many causes, ultimate stage of “burned-out” disease, untreated weakness may become fixed in relation to muscle atrophy.34


Integrated and useful content
I was diagnosed with Myasthenia Gravis in 2014, 2 years after they removed a tumor on my thymus. It’s been an okay road. Had one flare which put me in the hospital 4 years ago and this previous year I have been in and out of the hospital trying to see which treatment can get me back feeling 100% again. Pyridostigmine bromide just makes me nauseous all day and didn’t help my droopy eye or swallowing so I need to try something else which was natural herbs recommendation from multivitamincare. org This herbal treatment has successfully cure my MG and am 100% free ,I completed the herbal treatment program last year December and am very delighted i came across their website.it starts in the eyes and face then bulgar muscles , without treatment or the proper dosages of treatment it goes on to my limbs and then neck and breathing muscles but I can happily say that am free from MG after taking my chances to try natural herbs ,my neurologist was surprise after my result of being totally cured with herbal cure from the org.
I had myasthenia gravis since 2017. I got medically discharged out of the Army, a job I loved well. My heart had 5 myasthenic crises, 3 being severe enough to be ventilated or require CPAP (continuous positive airway pressure) helmet. I have had countless plasma exchanges as my veins are bad. I also needed Hickman lines inserted. I have been on azathioprine, mycophenolate (CellCept), methotrexate and none have worked. I’m currently done with my herbal remedy I purchase from Multivitamin herbal cure “ which has totally cured my condition with a surprise after almost 6 months of their usage, I was discouraged and never thought I would be myasthenia gravis (MG) free ,to me the best to get rid of this condition is multivitamincare. org treatment because all medications I used never worked include mycophenolate (CellCept).
I am a retired navy veteran and was diagnosed with myasthenia gravis in September 2020. After a bout of left ear infection and TMD (Temporomandibular disorder), symptoms persisting were eyelid drooping, slurred speech, drooling and overall muscle weakness in the left hand grip, increased fatigue and unsteady walking. I’m now taking www. madibaherbalcenter . com herbal cure (3 months) and I have been receiving a great improvement since I started the remedy, I find joy in being able to go out by myself and catch a movie. “It’s been wonderful, and it’s been life-changing.