Article topic: McArdle’s Disease
Author: Shahd Aktham Etoom
Editor: Ahmad Abuaisheh.
Reviewer: Ethar Hazaimeh
Keywords: McArdle disease (MD), glycogen storage disease type V (GSDV), glycogen phosphorylase (PYGM), creatine kinase (CK).
Synonyms: Glycogenosis Type V )GSDV(, McArdle Disease, Muscle Glycogen Phosphorylase Deficiency, Myophosphorylase Deficiency, PYGM Deficiency.
Abstract
Glycogen storage diseases (GSDs) are faulty hereditary glycogen metabolism conditions, characterized by deposits or atypical forms of glycogen in the liver, muscle, and brain tissues. This inborn metabolic mistake offers a model for comprehending the significance of glycogen in muscle function and the compensatory adaptations that arise in response to decreased glycogenolysis since muscle glycogen is an essential fuel for muscle during exercise. They are regarded as uncommon diseases because of their extremely low frequency.
Exercise intolerance, the second wind phenomenon, and elevated serum creatine kinase activity are the hallmarks of glycogen storage disease type V (GSDV), also known as McArdle disease. In the population that is affected by this disease, we recapitulate PYGM mutations. It has long been believed that McArdle’s illness is a metabolic myopathy brought on by a loss of expression of the muscle isoform of glycogen phosphorylase (PYGM).
Overview
The kind of glucose that is stored in the body, called glycogen, is mostly produced from consumed carbs. Multiple enzymes must work in concert to break down glycogen for energy; GSDs are caused by a flaw in one of these enzymes.
Depending on the type of cell, phosphorylase is an enzyme that is encoded by one of three genes; muscle (PYGM gene), liver (PYGL gene), or brain (PYGB gene). The most prevalent muscle GSD, GSDV, is caused by a lack of the muscle-specific isoform of glycogen phosphorylase (myophosphorylase)1.
A lack of this enzyme prevents the conversion of muscle glycogen into glucose-1-phosphate, preventing glycogenolysis from proceeding. However, due to the ability of muscle fibers to absorb blood glucose and convert it to glucose-6-phosphate after the metabolic block, glycolysis is only partially inhibited in GSDV.2
Individuals with GSDV experience muscle fatigue and pain, tachypnea, and tachycardia very soon after beginning physical activity and throughout all intense activities because skeletal muscle predominantly uses anaerobic energy for the first few minutes as it transitions from rest to activity, as well as throughout more intense activities. If these warning symptoms go unnoticed, a muscular contracture could develop very quickly and cause rhabdomyolysis.
A condition known as “second wind”, specific to GSDV, often happens 6 to 10 minutes into physical exertion, it was first recognized by Pearson et al. (1961) and signifies a noticeably increased capacity for physical exertion, such that formerly exhausting exercise is now more easily endured.3
Second wind is characterized by a sharp decline in heart rate, typically by 20 to 50 beats per minute, a marked improvement in the ability to work the muscle to extract oxygen and substrates from arterial blood, a significant drop in perceived exertion, and frequently a reduction in ventilation and breathing effort.4
Clinical overview
The key characteristic of GSDV is “intolerance to physical exercise”. Exercise, a type of physical activity, is planned and regulated, making it simpler to control. Activities of daily living (ADL) on the other hand, are frequently spontaneous, and symptoms might appear seconds to minutes after initiating them. Many people only obtain a diagnosis as a result of an incidental discovery of high serum creatine kinase(CK), despite having a long history of activity restrictions. Muscular tiredness and exercise-induced muscle pain are the predominant symptoms of pain and premature physical activity intolerance, and 30% of GSDV patients experience chronic pain.16
Epidemiology
The difference between the prevalence calculated using genetic information and the prevalence calculated using diagnosed cases may be caused by the typical delay in accurate diagnosis. Based on genetic data, the prevalence of GSDV in the Dallas/Fort Worth region of the USA is predicted to be 1/100,000.17
GSDV has an autosomal recessive inheritance pattern. The following gender ratios of males to females have been noted in the GSDV cohorts: Italy 65:35, Spain 55:45, and the UK 50:50.
In an autosomal recessive disease, equality is predicted. Given that men are more likely to engage in physically demanding leisure and employment activities, areas where affected men predominate may reflect gender-related reporting bias.18
Pathogenesis
Adenosine triphosphate (ATP), which is produced by skeletal muscles, is produced by three main metabolic processes:
(1) Oxidative phosphorylation
(2) Glycolysis
(3) Adenylate kinase and creatine kinase (CK) reactions.
The rate of ATP resynthesis must properly match the rate of consumption since the amount of ATP stored in skeletal muscle is limited and would be exhausted in a few seconds of sprinting if not renewed. Compared to glycolysis and adenylate kinase/CK activities, oxidative phosphorylation (aerobic ATP generation) produces more energy, although these anaerobic mechanisms can be engaged more quickly.19
The two types of exercise:
Aerobic exercise
It includes walking, gentle swimming, cycling, and jogging. Skeletal muscle used different types of fuel during aerobic activity depending on the type, intensity, and duration of the exercise, physical condition, and dietary habits. Since GSDV patients may tolerate aerobic exercise better and it favors the consumption of blood-borne substrates like fatty acids, it is useful as a treatment regimen.20
“Anaerobic” exercise
It is intense and cannot be continued (e.g., weightlifting or 100-meter dash). Normally, myophosphorylase converts glycogen to glucose during anaerobic exercise, which enters the glycolytic pathway and creates ATP “anaerobically” (or with no need for oxygen).20
Any workout has an anaerobic phase that lasts for the first few minutes. Muscle employs a variety of fuel sources, including anaerobic glycolysis, blood glucose, muscle glycogen, and aerobic glycolysis, followed by fatty acid oxidation, depending on the extent and duration of the exercise.20
Blood-free fatty acids are the body’s main source of energy when at rest. In the mitochondrial beta-oxidation route, these molecules are oxidized to create acetyl-CoA, which is then processed through the citric acid cycle and mitochondrial respiratory chain to produce ATP.20
Clinical Presentation and Complications
A metabolic myopathy known as McArdle’s disease typically exhibits exercise intolerance as rapid onset tiredness, myalgia, and cramping in working muscles. 20 Some older patients experience muscle atrophy and weakness.15
Although this clinical picture is typically conventional, some patients may show mild or severe manifestations. For instance, in certain cases, the disease manifests at a very young age with hypotonia, generalized muscle weakness, and progressive respiratory failure.22
85% of patients experience symptoms before the age of 10, however, the intensity of the symptoms varies, and about 50% of those people were diagnosed for the first time after the age of 30.21 These symptoms are frequently brought on by persistent, powerful “aerobic” exercise (such as jogging or climbing stairs) or isometric exercise (such as hauling weights), and are typically eased by rest, it can happen to any skeletal muscle.21
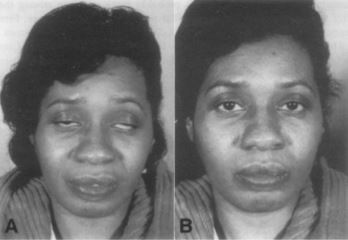
Additionally, atypical manifestations have been reported, including issues with mastication, dysphagia, oral motor function (which appear to be more common in younger individuals)23, spontaneous compartment syndrome,24, 25, and acute contracture of the posterior neck muscles.26
Continuing to work out while experiencing intense discomfort, however, could lead to myoglobinuria and muscle damage (rhabdomyolysis). About 50% of people get myoglobinuria from rhabdomyolysis after vigorous exercise; few people really experience acute renal failure, despite the danger. Despite the fact that renal failure is almost always treatable, it needs to be treated immediately.29 It is evident that having a history of dark urine may prevent GSDV misdiagnosis and consequences. 26, 30
Diagnosis/Workup
GSDV’s clinical symptoms often appear in childhood.29, 31 In GSDV, basal CK is frequently elevated. In young toddlers, the ‘second-wind’ phenomena might not be as obvious.32
In short, it is difficult for clinical care experts to diagnose these individuals because the differential diagnosis for this disease is very complex. Pediatricians should explore these pathologies even though they are rare disorders because the initial presentation typically occurs before the age of 10, and today the first line of defense is frequently genetic testing. When making a differential diagnosis for adults, the patient’s self-report of “second wind” or a flat lactate response with an increase in ammonia levels following a non-ischemic forearm exercise test may be helpful.33
The first presentation of symptoms to a general practitioner (GP) often occurs before the age of 10 years; nevertheless, the median age of a correct diagnosis is 33 years old. Children are frequently misdiagnosed by GPs as being “lazy or unfit” (51% of patients) or experiencing “growing pains” (44%), two of the most frequently reported misdiagnoses.33
Males and females are misdiagnosed equally, with the exception of psychological problems, when females are misdiagnosed six times more often than males.33 With the increasing use of genetic testing, the misdiagnosis of neuromuscular illnesses is projected to decline.34
Clinical symptoms, high serum creatine kinase levels, and a lack of lactate rising during anaerobic activity are used to make the diagnosis, which is supported by genetic testing and/or muscle biopsy.35 A blood test to assess basal CK in GSDV is helpful as a screening test because it is nearly certain to be highly elevated indicating the need for additional investigations.33
The absence of genotype-phenotype association in GSDV is explained by a recent article. Regardless of the kind of mutation (missense, nonsense, deletion, insertion, splicing, etc.), the majority of the research participants lacked myophosphorylase function.34 Clinical severity, PYGM genotype, and muscle biochemical tests do not appear to be significantly correlated.35
After taking a careful clinical history, observing elevated serum CK levels, and employing minimally invasive techniques based on molecular analysis of the PYGM gene on deoxyribonucleic acid (DNA) isolated from peripheral blood samples, the diagnosis of GSD V can be made. Clinical molecular genetic testing is provided through whole exome sequencing, NGS panels with PYGM (for all GSDs or for rhabdomyolysis, for example), and Sanger sequencing of PYGM (WES). For a diagnosis to be confirmed, two pathogenic mutations must be found in suspected patients.
Single-gene Sanger sequencing, myopathy or rhabdomyolysis gene panels, or WES with peripheral blood samples can all be used for genetic testing.36,37 Targeted mutation analysis can be used when a proband has known disease-causing familial mutations.21, 38, 39, 40
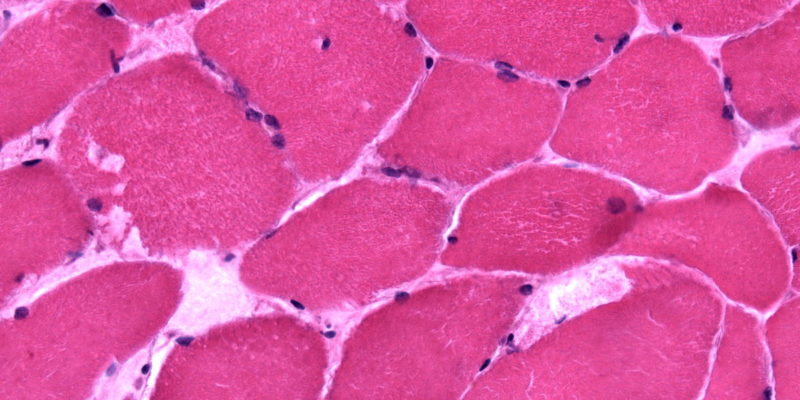
When variations of uncertain significance (VUS) are found, functional exercise tests like the cycle test, non-ischemic forearm tests, and muscle biopsies for the activity of the muscle phosphorylase enzyme may be helpful.36, 37 Exercise tests may not be definitive when VUS is present, but a biochemical or histochemical analysis of the muscle biopsy tissue may reveal an enzymatic abnormality.21,41,42 Muscle biopsy samples from people with GSDV typically exhibit abnormally elevated amounts of subsarcolemmal glycogen buildup (3 to 5 times the normal range).40
Enzymatic analysis of skeletal muscle biopsies, which reveals undetectable or extremely low levels of myophosphorylase activity, confirms the diagnosis of GSDV.38,39 In GSDV samples, myosin-heavy-chain labeling did not reveal any distinctive muscle fiber type distribution patterns.43
With the help of both cycle ergometry and walking tests, the value of the “second wind” as a diagnostic sign has been investigated.44,45 By monitoring heart rate and subjective muscular discomfort every minute, the 12-minute walk test can be administered with ease in a clinical setting. It’s vital to keep in mind that in very young patients with GSDV, the “second-wind” phenomena might not be readily apparent,32 and some people could need assistance to recognize the “second wind”46, 47, 48, 49.
Forearm exercise test
Patients with muscle GSD have done the forearm exercise test. A non-ischemic variant of the test is preferable, according to studies in GSDV, as ischemia in many individuals causes cramping and agony along with the possibility of rhabdomyolysis.50
Patients with GSDV exhibit a flat lactate response even when forearm muscles are not ischemic due to cuff pressure. As a result, a non-ischemic test is advocated for the diagnosis of GSDV, and patients should be told to cease the test if they feel any pain in their muscles.51,52
Lab testing and imaging
In a group of 256 people with GSDV, the median basal CK elevation was 2643 IU/L, and just 18 people (7%) had normal values ( 200 IU/L) reported.49 CK levels can be very high during episodes of rhabdomyolysis, sometimes exceeding 100,000 IU/L. When starting new medications or testing without an accident, it’s crucial to establish a baseline CK level. Patients will be better able to control their disease and avert dangerous episodes thanks to this.
The liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are virtually invariably high when CK levels are over the thousands. This is a normal dispersion of sarcoplasmic muscle transaminases brought on by fiber damage rather than a symptom of liver disease. Further research may be necessary if ALT and AST levels are markedly high compared to the hyperCKemia, or if bilirubin or alkaline phosphatase (ALP) levels are markedly elevated. Myoglobin may be present if pee test strips reveal blood (hemoglobin) or protein. Urate levels are frequently elevated in GSDV as a result of excessive rises in blood ammonia, inosine, and hypoxanthine brought on by the rapid breakdown of muscle purine nucleotides, which act as building blocks for uric acid synthesis. This could result in the emergence of hyperuricemia and gout.
Additionally, kidney stones might form as a result of elevated urate levels. Due to the higher frequency of diabetes and coronary artery disease in this population, routine HbA1c testing and lipid profile analysis are recommended [Quinlivan, R; unpublished data].73
Standard muscle MRI evaluation often reveals no fatty infiltration or muscular edema. Lower leg and shoulder girdle muscles may become fatty throughout a long-term illness course. Therefore, illness distinction or therapy monitoring is not possible with routine clinical MRI. Only C13 and P31 magnetic resonance spectroscopy has been able to non-invasively demonstrate abnormal glycogen buildup and metabolism in McArdle disease to date.60, 61, 62, 63, 64
New (quantitative) MR imaging techniques must be examined in order to take part in an early disease evaluation. Muscle diffusion tensor imaging (mDTI) can assess the diffusion characteristics of water molecules in muscle tissue to reveal information about muscular microstructure.65, 66, 67
Management & Treatment
The aim of treatment should be to lessen episodes of muscular breakdown, which may lead to hospital admissions for serious and perhaps fatal conditions such as rhabdomyolysis, ARF, and ACS.
Patients who present for the first time later in life are also more likely to have adopted unhealthy lifestyles and lost fitness. They may find it difficult to find their “second wind” as a result, and they may experience chronic discomfort and weariness as well as a low quality of life.68
The majority of patients should have their annual assessment. To establish physical fitness/functional capacity and the ability of patients with GSD V to use the “second wind,” an assessment may include a functional exercise test. This might either be a 12-minute walk test68 or a 12-minute cycle test46. A blood draw to evaluate basal CK, urate, HbA1c, and lipid profiles is advised once a year because of the greater prevalence of GSD V compared to the general population of gout (8.5: 5%), diabetes (6.7: 4.8%), and myocardial infarction (11: 4%) .49
Guidance of day-to-day management
To increase physical activity tolerance and lessen muscle injury, patients must be aware of how to get a “second wind.” The muscles that are being worked on are what cause the “second-wind” phenomenon. Patients with GSD V must move slowly and be aware of the following symptoms while beginning an activity: muscle tiredness or pain, rate of perceived exertion, increased heart rate, and increased breathing effort. Patients are recommended to slow down (lower exertion) or stop until symptoms go away if any of these signals appear.69
The maximum amount of oxygen that can be used during physical exercise “VO2 max” is the best indicator of aerobic conditioning. Exercise that is “appropriate” for you can raise your VO2 max. only low to moderate-intensity work, ideally lasting at least 20 minutes, can be performed by people with muscle GSDs 2-4 times per week. Strength training can increase muscle mass, reduce the severity class, lower the baseline CK, and prevent fixed muscle weakness in the case of GSDV. It is crucial to follow the rules exactly to reduce the chance of contractures. The basic idea is to warm up all of your muscles to get them into a “second wind”, then perform sets of relatively few repetitions utilizing a circuit training framework, rotating on different machines to allow at least three minutes between bouts of rest.70, 44, 71, 72
A revised and updated systematic review of dietary and pharmaceutical interventions for GSDV published in the Cochrane Database found no benefit from high-dose oral ribose, a fat-rich diet, glucagon, verapamil, vitamin B6, a high-protein diet, branched-chain amino acid supplementation, dantrolene sodium, a high-dose of creatine, an intravenous course of gentamicin, a ketogenic diet, or intralipid Oral sucrose, a diet high in carbohydrates, ramipril, and low-dose creatine were treatments that provided some improvement.75
Surgery
The primary common factor is the generalized or focal failure of blood glucose, the easily accessible energy source that is given to skeletal muscle, whether due to inadequate supply or excessive consumption. Thus, it is advised that patients consult with the anesthetist in person before significant surgeries to go over the safety measures.
Prognosis
In general, this condition does not impact the affected person’s life expectancy.22
Conclusion
Future treatment trials must work with several centers and utilize consistent assessment techniques due to the rarity of GSDV.20
There is currently no known cure for McArdle disease, although treatments often focus on managing the disease’s symptoms and avoiding strenuous exertion. Adjuvant therapy is based on executing a controlled physical activity to increase glucose intake relative to the exercise periods and muscle mitochondrial oxidation capacities.21,76,2
Affected people benefit from moderate-intensity aerobic exercise to improve their cardiorespiratory fitness and muscle oxidative capacity. Examples of such exercise are biking, strolling, and brisk walking. Pre-exercise consumption of sports drinks with simple carbohydrates increases exercise tolerance and may prevent rhabdomyolysis brought on by exercise.20