Topic: Seizures and Epilepsy: A Comprehensive Clinical Overview
Author: Ayah Alatoom.
Editors: Ghina Mohamad Kaskas, Ihda Bani Khalaf
Reviewer: Ethar Hazaimeh
Keywords: Seizure, epilepsy, status epilepticus, anti-seizure medications
Overview
Seizures and epilepsy represent a major aspect of neurological practice, carrying significant clinical, social, and prognostic implications. A seizure is defined as a transient occurrence of signs or symptoms resulting from abnormal, excessive, or synchronous neuronal activity in the brain. While a single seizure may occur in the setting of an acute insult, epilepsy is a chronic disorder characterized by a predisposition to recurrent, unprovoked seizures.
Seizures may be focal, arising from a localized region of the cortex, or generalized, involving widespread networks from onset. They manifest with a wide spectrum of clinical features, ranging from subtle sensory changes to dramatic motor convulsions or alterations in awareness. Classification systems, such as those developed by the International League Against Epilepsy (ILAE), provide a framework for accurate diagnosis and guide treatment decisions.
Management of seizures and epilepsy requires a comprehensive approach that balances acute interventions with long-term therapy, whether through antiseizure medications or other non-pharmacological options. Equally important are the broader consequences of epilepsy, which extend beyond seizure control and require early recognition and management integral to holistic care.
Epidemiology
The point prevalence of epilepsy is between 4-10 per 1,000 persons, while the annual incidence is 62 per 100,000, with 71% being classified as focal, 11% as generalized, and 18% being non-classified. Epileptic seizures account for 1% of hospital admissions and 3% of emergency department visits. The incidence of epilepsy varies between age groups, with the highest rates in young children and older adults. In children, the incidence is highest in the first year of life and then declines gradually. Generalized epilepsy is more often diagnosed in younger age groups, being almost exclusive to those below age 25. On the other hand, focal epilepsy has a higher incidence in older age groups, whereas non-classified epilepsy remains relatively flat across all age groups (1).
Definitions
A seizure is an excessive, abnormal, and/or hypersynchronous neuronal firing leading to transient neurological symptoms.
Acute symptomatic seizures are seizures occurring in close temporal relationship with a CNS insult, whether metabolic, inflammatory, toxic, or infectious. The use of the term acute symptomatic seizure is recommended instead of provoked seizure, reactive seizure, or situation-related seizure. They are not considered epilepsy, because they are a direct and immediate consequence of a reversible condition. The “close temporal association” window depends on the cause. Seizures are considered acute symptomatic if they happen within 7 days of cerebrovascular disease, anoxic encephalopathy, TBI, including intracranial surgery, CNS infection, multiple sclerosis relapse, or as a first symptom of a relapse, and during the activation of an autoimmune disease. More examples of acute symptomatic seizures include during a disturbance by illicit drug use, alcohol intoxication, or withdrawal (2).
Reflex seizures are seizures consistently triggered by specific stimuli or activities that lower the seizure threshold. Unlike spontaneous seizures, reflex seizures have identifiable and repeatable external (or internal cognitive) precipitants (3).
Unprovoked seizures are defined as seizures in the absence of an identifiable cause or beyond the specified interval of a CNS insult, beyond the interval for an acute symptomatic seizure (2).
To describe events, clinical features, and Electroencephalogram (EEG) findings and signs in relation to seizure, we use the terms ictal, inter-ictal, and post-ictal. Derived from the Latin word “ictus”, meaning “strike” or “sudden attack”, ictal refers to happening during a seizure, inter-ictal in between seizures, and post-ictal after a seizure (4).
Epilepsy is a chronic neurological disease in which the brain demonstrates a pathological, enduring tendency to have recurrent seizures. It is defined as two unprovoked or reflex seizures occurring at least 24 hours apart, or one unprovoked or reflex seizure with a high risk of subsequent seizures –a high risk being 60% or more over the course of 10 years– such as a single seizure within less than a month of stroke or presence of a structural etiology and epileptiform EEG. The definition of epilepsy can also be applied to the diagnosis of an epilepsy syndrome –it makes little sense to say someone has an epilepsy syndrome but not epilepsy (3).
Resolved epilepsy is used when a person no longer has the disease, but the term does not guarantee that seizures will not recur. It is defined as the absence of recurring seizures for 10 years with at least 5 years being without taking anti-seizure medications (ASM), or an individual who was diagnosed with an age dependent age-dependent epilepsy syndrome who is now past the applicable age for that syndrome (3).
Drug-resistant epilepsy (DRE) is defined as the failure of adequate trials of two tolerated, appropriately chosen, and used anti-seizure medication (ASM) schedules, used either as monotherapies or in combination, to achieve sustained seizure freedom. Sustained seizure freedom is typically defined as freedom from seizures for a minimum of 12 months, or for a period three times the longest pre-intervention inter-seizure interval, whichever is longer (5).
Sudden unexpected death in epilepsy (SUDEP) is defined as death in a patient with epilepsy that is not due to trauma, drowning, status epilepticus, or other known causes, but for which there is often evidence of an associated seizure and represents a leading cause of death in patients with epilepsy (6).
Status epilepticus (SE) is an abnormally prolonged seizure lasting more than a specific duration, or a series of seizures that happen in rapid succession to one another without full neurological recovery in the inter-ictal state, and if not terminated, can produce long-term consequences. The specific duration of time after which a seizure is considered status epilepticus depends on the seizure type. For tonic-clonic, it’s 5 minutes, for focal seizures with impaired awareness, it’s 10 minutes, and for absence seizures, it’s 10-15 minutes (7).
Febrile seizures are seizures which happen in children between the ages of 6 months to 5 years, associated with fever (≥ 38°C/100.4°F), and in the absence of a CNS infection, metabolic abnormalities, or a history of afebrile seizures. They may be classified based on clinical features into simple febrile seizures, complex febrile seizures, and febrile status epilepticus (8).
Etiology
Epilepsy Causes and Risk Factors
The most recent classification of the epilepsies provides a new framework at three levels and highlights the importance of considering etiology at each level: seizure type, epilepsy type, and epilepsy syndrome (9). The etiological categories include structural, genetic, infectious, metabolic, immune, and unknown.
Structural brain lesions are associated with focal epilepsy and are divided into six main disease categories: hippocampal sclerosis, brain tumors, malformations of cortical development, vascular malformations, glial scarring, including following stroke and traumatic brain injury (TBI), and brain inflammation (10).
Hippocampal sclerosis, constituting the largest category, is a heterogeneous disorder and is the result of a complex interplay between genetic predisposition and environmental insults like prolonged febrile seizures, TBI, or infectious causes.
Brain tumors of glioneuronal composition are of particular interest in epilepsy. Such tumors are often of low malignancy, occur in childhood, and arise mostly in the temporal lobe, thus presenting with seizures. Examples include ganglioglioma and dysembryoplastic neuroepithelial tumors.
The most common brain malformation associated with epilepsy is focal cortical dysplasia (FCD). It often presents as Drug-resistant epilepsy (DRE) in early childhood, and the ILAE classification of FCD includes type I with abnormal cortical dyslamination, type II with cortical dyslamination and dysmorphic neurons, and type III with another principal lesion.
Vascular malformations such as arteriovenous malformations and cavernous hemangiomas (Cavernoma) are associated with seizures. However, to generate seizures, they often require a secondary phenomenon like hemorrhage or hemosiderin deposition, and therefore, presentation often comes later in life.
Glial scars could result from TBI, perinatal injury like hypoxic-ischemic encephalopathy, hemorrhagic injury, or strokes. Interestingly, scars resulting from neurological surgeries do not cause seizures.
Genetic etiologies of epilepsy include mutations affecting ion channels functioning –channelopathies. These could be sodium channel genes like SCN1A mutations associated with Dravet syndrome and genetic epilepsy with febrile seizures plus (GEFS+), or gene mutations affecting potassium, chloride, or calcium channels interfering with neuronal firing. Genetic mutations could also affect neurotransmitter receptors, such as mutations in the GABAA receptor subunit genes GABRA1, GABRB3, and GABRG2, causing generalized epilepsy, synaptic vesicles, or the mTOR pathway, like the TSC1 and TSC2 gene mutations in tuberous sclerosis complex. In addition, mutations could disrupt cortical development, migration, and organization, ending in structural brain malformations, or interfere with energy metabolism when occurring in mitochondrial and metabolic genes. The identification of genetic etiology guides treatment through precision therapies like the ketogenic diet for GLUT-1 deficiency syndrome, avoidance of sodium channel blockers due to SCN1A loss of function, or conversely preferring the use of sodium channel blockers in SCN8A gain of function epilepsies, and using mTOR inhibitors for tuberous sclerosis-associated epilepsy, like everolimus.
Cerebral infections are a particularly important cause of epilepsy in developing countries. The causative agent could be either viral, bacterial, fungal, or parasitic. In sub-Saharan Africa, 26% of epilepsies are caused by infection, Malaria, Toxocara Canis, Toxoplasma Gondii, Onchocerca Volvulus, and Taenia Solium are among the most common causes. Neurocysticercosis is the most common parasitic infection causing epilepsy worldwide, and in endemic countries, around 30% of epilepsy cases are attributed to it. Bacterial meningitis carries a high risk of neurological sequelae. Tuberculous meningitis, often with a background of HIV infection, may also lead to epilepsy.
Metabolic causes of epilepsy are mostly made up of inborn errors of metabolism. These disorders involve defective proteins and metabolic abnormalities in the brain. It could be from failure of brain metabolism, deficiencies in important vitamins or cofactors, accumulation of abnormal storage materials or toxins, or disruption of the neurotransmitter systems. Examples include phenylketonuria and organic acidemias.
Immune causes include autoimmune encephalitis with antibodies directed against surface antigens like NMDA, AMPA, GABA-B receptors, or mGluR5. Many of which have been linked to malignancies like ovarian cancer, small-cell lung carcinoma, and Hodgkin lymphoma. Antibodies to intracellular antigens could also result in seizures, such as the paraneoplastic antibodies (anti-Hu, Ma, CRMP2, and amphiphysin antibodies) and anti-GAD65 antibodies. Rasmussen encephalitis is another important immunological cause of epilepsy, with a progressive inflammatory process affecting one hemisphere of the brain.
Dementias are the most common neurodegenerative disorders, among which Alzheimer’s disease is the most common. Early-onset Alzheimer’s disease carries a higher risk of epilepsy and is more likely to be of a genetic cause. Down syndrome has also been linked to epilepsy as a genetic condition with a risk for neurodegenerative diseases. In children with Down syndrome, West syndrome is the most common cause of epilepsy. But in the context of epilepsy and neurodegenerative disorders, adults with Down syndrome often develop Alzheimer’s disease, and the risk of epilepsy substantially increases after the onset of Alzheimer’s.
Seizure Triggers
Triggers are stimuli that could ignite a seizure in people with epilepsy. They could refer to reflex seizure triggers or non-reflex seizure triggers.
In epileptic patients without reflex seizures, these triggers are non-specific as they do not consistently and exclusively lead to seizures, working by lowering the seizure threshold and increasing the likelihood that a seizure may occur. Such examples include excessive physical exertion, mental stress, sleep deprivation, alcohol, flashing lights, hormonal changes, problems with medications in people known to have epilepsy, such as non-adherence, issues with drug dosing, introducing a new medication, or drug interactions.
Triggers of reflex seizures consistently and objectively induce seizures in patients with reflex epilepsy syndromes, which are relatively uncommon, representing approximately 5%–7% of people with epilepsy. There’s a notion that they manifest the functional brain regions generating them, as the trigger and ictal semiology are strikingly similar. For example, music triggers reflex seizures with an auditory aura featuring temporo-lateral epilepsies, though it remains unclear whether these seizures are caused by the emotional response to music or the sound itself, as they’re probably linked to a complex network between hearing and emotions. Nonetheless, pure tones have also been reported to trigger seizures. Other examples of reflex seizure triggers include somatosensory stimuli like tapping, rubbing, or toothbrushing, photic stimulation or eye closure, and high-level complex stimuli like reading, writing, calculating, or praxis (11,12).
Fever is the most common seizure trigger in children, manifesting as febrile seizures. And although febrile seizures do not meet the definition of epilepsy, complex febrile seizures and a particular type of genetic epilepsy, known as febrile seizure, plus a phenotype of Generalized Epilepsy with Febrile Seizures Plus (GEFS+), are associated with an increased risk of developing epilepsy at a later stage.
Causes of Acute Symptomatic Seizures
Common causes of acute symptomatic seizures include structural acute CNS injury (ischemic or hemorrhagic stroke, traumatic brain injury), CNS infections, acute demyelination, metabolic and systemic disturbances (hypoglycemia, hyperglycemia, hyponatremia, hypernatremia, hypocalcemia, hypomagnesemia, uremia, hepatic encephalopathy), toxic causes (drug intoxication with isoniazid, cocaine, theophylline, or drug withdrawal such as from alcohol, benzodiazepines, barbiturates), posterior reversible encephalopathy syndrome (PRES), and hypoxic–ischemic encephalopathy (2).
Causes of status epilepticus
Most cases of status epilepticus (SE) are caused by acute processes such as a CNS infection, electrolyte and metabolic derangements (hypoglycemia, hyponatremia, hypocalcemia, hepatic encephalopathy, and inborn errors of metabolism in children), cerebrovascular accidents, head trauma, drug toxicity (tricyclic antidepressants, isoniazid) or withdrawal (alcohol, benzodiazepines, barbiturates), hypoxia, autoimmune disorders, and hypertensive emergencies. Chronic conditions can also result in SE; they include an established epilepsy diagnosis with breakthrough seizures or a low dose of anti-seizure medication, either from non-compliance or miscalculation. Other chronic processes, like tumors and late-stage neurodegenerative diseases, have been documented to cause SE as well. In children, febrile SE is the most common cause, and most patients have no history of a prior seizure (13).
Pathophysiology
Seizures happen when there’s abnormal, excessive, synchronous electrical activity of primarily cortical neurons. Everyone has an inherent susceptibility to having a seizure; what influences who does and who doesn’t is the concept of a seizure threshold. This threshold is influenced by genetic, metabolic, toxic, inflammatory, infectious, structural factors, electrolyte disturbances, and even sleep patterns. If these influences push a person past the threshold, a seizure is generated.
When susceptible neurons become hyperexcitable, they recruit adjacent ones; the result is synchronized discharges by a progressively larger group of neurons. This recruitment process could happen through local as well as global tracts like the corpus callosum.
An imbalance between inhibitory and excitatory factors is key for this; an excess in excitation, mediated primarily by glutamate, combined with a deficiency in inhibition by GABA, is central in initiating these abnormal discharges. An electrophysiological process hallmarking this is paroxysmal depolarization shifts (PDS); excessive, synchronized excitatory input produces prolonged depolarization exceeding the normal excitatory postsynaptic potential (EPSP). This is followed by a high frequency of repetitive action potentials occurring in rapid succession, and finally, afterhyperpolarization as potassium efflux and inhibitory inputs restore the membrane potential. In a seizure-prone network, large populations of neurons will undergo PDS simultaneously, producing the hypersynchronous discharges seen in seizures (14).
Classification
Classification of seizures
In The International League Against Epilepsy (ILAE) revised operational classification of seizure types (figure 1), seizures are classified according to localization of the abnormal neuronal activity at the onset of the seizure, then further subclassified based of the level of awareness and seizure symptoms (15).
Focal seizures originate from the networks of a single hemisphere, generalized seizures originate from both hemispheres, and if the onset is missed or obscured, the seizure is of unknown onset. Focal seizures can then be optionally subcategorized based on the level of awareness, whether the person is aware of the self and the environment during the seizure or not, regardless of his/her mobility. Focal aware seizures and focal impaired awareness seizures correspond to the old terms “partial simple seizures” and “partial complex seizures”, respectively. The other classifier of focal seizures is the symptoms at the onset of the seizure: motor and non-motor.
“Focal to bilateral tonic-clonic”, formerly termed “partial onset with secondary generalization”, is a special seizure type. Due to its commonality and importance, it has its own separate category, though it reflects the propagation pattern of the seizures rather than a unitary seizure type.
In generalized seizures, seizure activity begins in and rapidly engages bilateral networks, so awareness is usually impaired from onset. Therefore, further subdivisions are only by symptoms to motor and non-motor (absence) seizures. Generalized seizures may have asymmetrical symptoms, rendering it difficult to distinguish from focal-onset seizures.
When the determination of focal or generalized onset is not possible, the seizure is referred to as a seizure of unknown onset. This term is only a placeholder, not a characteristic of the seizure, as it comes from ignorance and allows future better classification and characterization of the seizure.
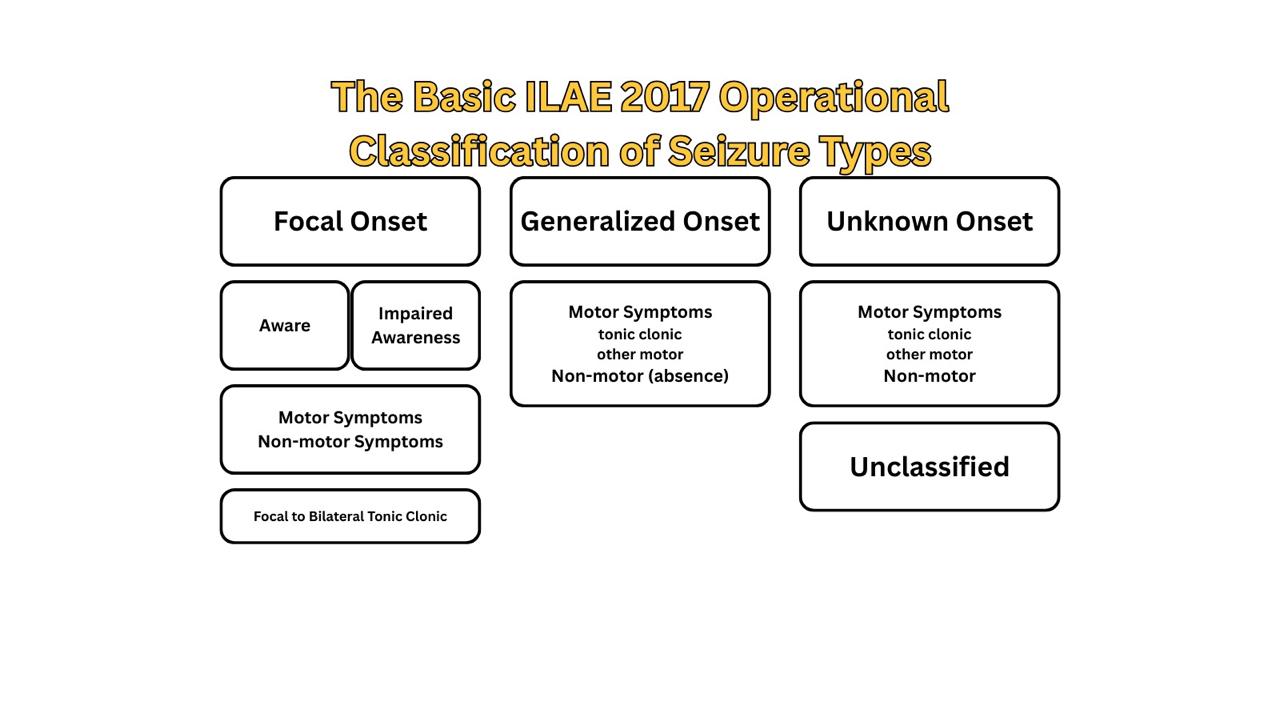
Figure 1: The basic ILAE 2017 operational classification of seizure types
Classification of epilepsy
The International League Against Epilepsy (ILAE) recommends a multilevel classification of epilepsy (Figure 2): seizure type, epilepsy type, and epilepsy syndrome. Alongside these three levels, the clinician should aim to establish an etiological diagnosis, as well as consider comorbidities for every patient with epilepsy at each stage of classification (9).
Determination of seizure type is the starting point for classifying epilepsy and is based on the previously mentioned seizure classification: focal-onset, generalized-onset, and unknown-onset seizures.
The second level is epilepsy type, which can be focal, generalized, combined generalized and focal, or unknown. The diagnosis of epilepsy type is made on clinical grounds and supported by EEG findings. Generalized epilepsy is diagnosed in patients with generalized-onset seizures, and they may have a range of seizure types from tonic-clonic to myoclonic or absence seizures. Focal epilepsy patients have focal-onset seizures, which include focal-to-bilateral tonic-clonic seizures as well, and the hemisphere from which the seizures are ignited should be determined. If a patient has both focal-onset and generalized-onset seizures, the epilepsy type is combined generalized and focal. Classical examples of this type of epilepsy are Dravet syndrome and Lennox-Gastaut syndrome.
For the third level, a diagnosis of an epilepsy syndrome is made, if possible, as the epilepsy type may be the final level of classification. An epilepsy syndrome incorporates a cluster of features: seizure type, EEG and imaging findings, age-dependent events and features, diurnal variations, and sometimes coexisting comorbidities and intellectual or psychiatric disorders. It is noteworthy to mention that there’s no official classification of epilepsy syndromes made by the ILAE. However, the epilepsydiagnosis.org website provides an excellent resource for navigating and understanding the parameters for diagnosis.
At each level of classification, etiology and comorbidities should be considered. An etiology of a person’s epilepsy could be structural, genetic, metabolic, immunological, infectious, or unknown. They’re not hierarchical, and epilepsy could be classified into more than one etiological category. Many epilepsies are associated with psychological, intellectual, behavioral, and other problems, varying in type and severity. Therefore, like etiology, comorbidities are assessed with every advance in classifying epilepsy, to enable early identification and proper management.
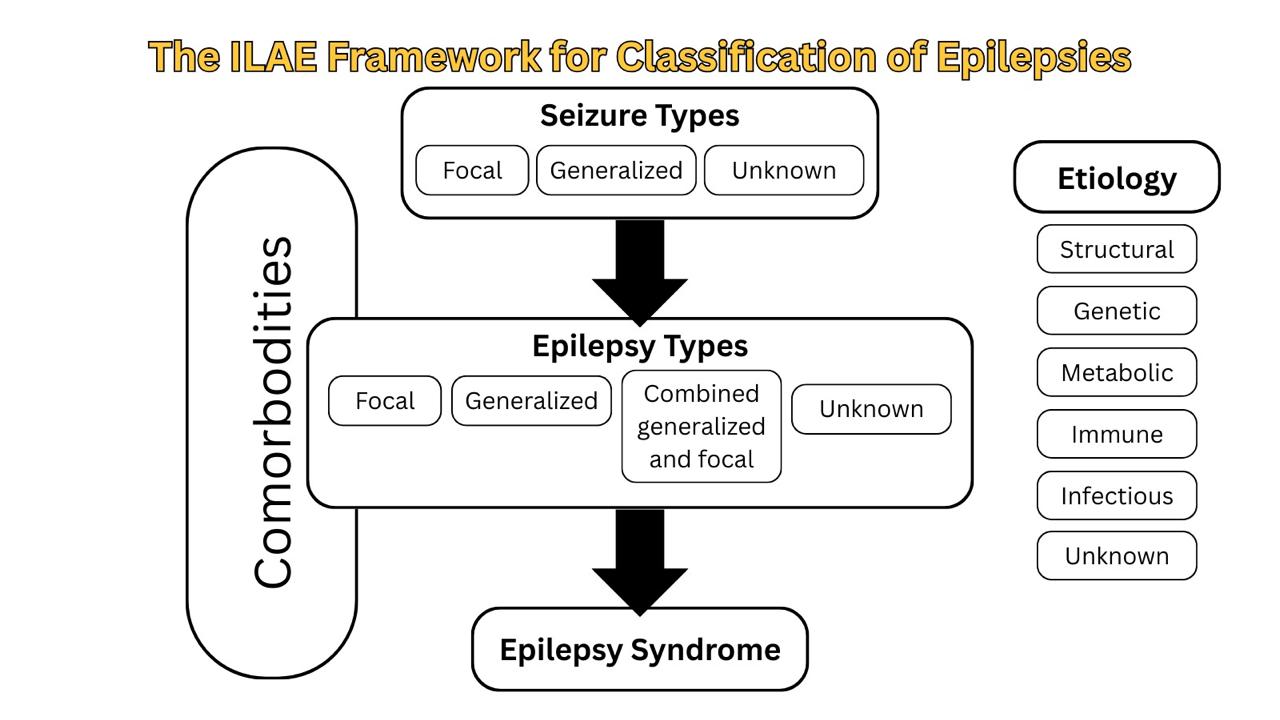
Figure 2: : The 2017 ILAE framework for classification of epilepsies
Classification of status epilepticus
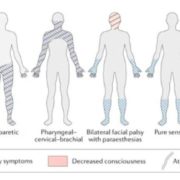
The ILAE Task Force proposed a stepwise classification into four main axes: semiology, etiology, EEG correlates, and patient’s age (Figure 3). It is acknowledged that categorization according to each axis is not always possible; still, a patient’s age and seizure semiology are possible to assess upon initial presentation (7).
Seizure semiology is the backbone of this classification, with two main criteria: presence or absence of prominent motor activity, and the degree of impaired consciousness. The subtypes of status epilepticus with prominent motor features are convulsive SE (CSE, synonym: tonic-clonic SE), myoclonic, focal motor, hyperkinetic, and tonic SE. Status epilepticus without prominent motor symptoms (i.e., nonconvulsive SE, NCSE) is further subdivided into NCSE with coma and NCSE without coma. NCSE without coma includes the generalized (typical absence, atypical absence, and myoclonic absence), focal (Without impairment of consciousness, with impairment of consciousness, and aphasic status), and unknown (includes autonomic status) subgroups (7).
SE is classified by etiology into known (acute, remote, progressive, SE in defined electroclinical syndromes) and unknown groups. Although there is no evidence-based EEG criteria for SE and there are no specific features, it remains indispensable for diagnosis. The following terms are recommended for use in describing the EEG patterns in SE: Location (generalized, lateralized, bilateral independent, multifocal), name of the pattern (periodic discharges, rhythmic delta activity or spike-and-wave/sharp-and-wave plus subtypes), morphology (sharpness, number of phases, absolute and relative amplitude, polarity), time-related features (prevalence, frequency, duration, daily pattern duration and index, onset and dynamics), modulation (stimulus-induced vs. spontaneous), and effect of intervention (medication) on EEG. Finally, SE can be classified according to the age group they occur into neonatal (0 to 30 days), infancy (1 month to 2 years), childhood (> 2 to 12 years), adolescence and adulthood (> 12 to 59 years), and elderly (≥ 60 years) (7).
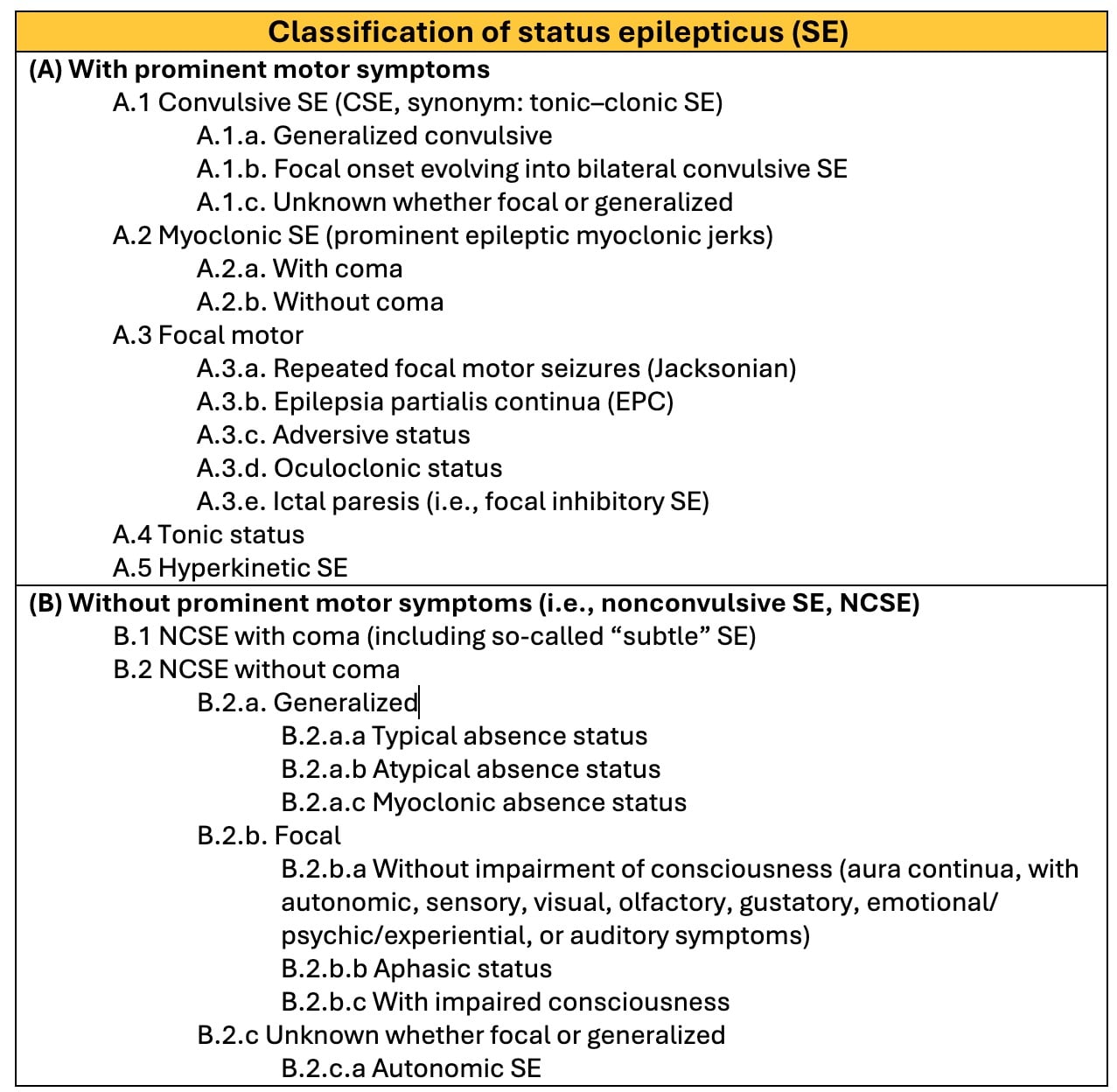
Figure 3: The ILAE classification of status epilepticus (SE)
Clinical Features
Seizure symptoms provide valuable clues for localization and lateralization of epileptic foci. These symptoms reflect the functional role of the brain region from which they arise. A solid understanding of brain physiology will help appreciate how the seizure semiology correlates with the underlying neural origin (16,17).
In between seizures, patients are usually asymptomatic. However, due to the sedative effects of some antiseizure medications, they may exhibit an overall decrease in alertness. Other times, the underlying neurological condition causing the seizure may lead to deterioration of mental functions and health (16,17).
Auras pinpoint the onset of the seizures but are still referred to as “prodromal”. Auras are a subjective seizure semiology –how the patient feels, preceding the objective observed signs of the seizure, and could be motor, sensory, or neuropsychiatric (16,17).
A post-ictal state lasts from minutes to hours, and it might include deep sleep, general fatigue, headache, confusion, residual transient neurological deficits, depending on the cerebral region, and sometimes Todd’s paralysis; weakness or paralysis of the muscles or limb that was involved in the seizure (16,17).
Focal-Onset Seizures
Focal-onset seizures may manifest with intact or impaired awareness, the latter most commonly originating from the temporal lobe. The main symptom could be motor, sensory, autonomic, or cognitive. Motor focal seizures could manifest with automatisms, atonic, myoclonic, clonic seizures, Jacksonian march, or hyperkinetic seizures. Sensory symptoms might be tingling sensations, numbness, visual, gustatory, olfactory, auditory hallucinations, or vestibular symptoms. Dyslexia, anomia, anterograde amnesia, and aphasia are examples of cognitive symptoms.
Specific symptoms of focal seizures help in localization, as they reflect the epileptogenic region of the brain. For example, simple movements like limb twitching originate in the primary motor cortex, abnormal taste sensation –dysgeusia, arises from the insula, and complex behavioral automatisms point towards the temporal lobe. More examples of the site of dysfunction according to seizure manifestation are in (Table 1).
Focal-to-bilateral onset seizures start with unilateral, focal symptoms then progress to a bilateral, generalized phase. In the generalized phase, the patient loses consciousness and may experience motor symptoms such as tonic-clonic seizures and/or bowel and bladder incontinence. The initial unilateral phase may go unnoticed, leading to a misdiagnosis of generalized-onset seizures.
A rare disorder called Epilepsia partialis continua is a prolonged, continuous focal aware seizure involving a particular body part like the hand, face, or arm. Seizures can persist for several days. The cause is unspecific but can be a structural damage to the cortex, such as stroke in adults, or a focal cerebral inflammatory process like viral encephalitis or Rasmussen encephalitis in children (13).
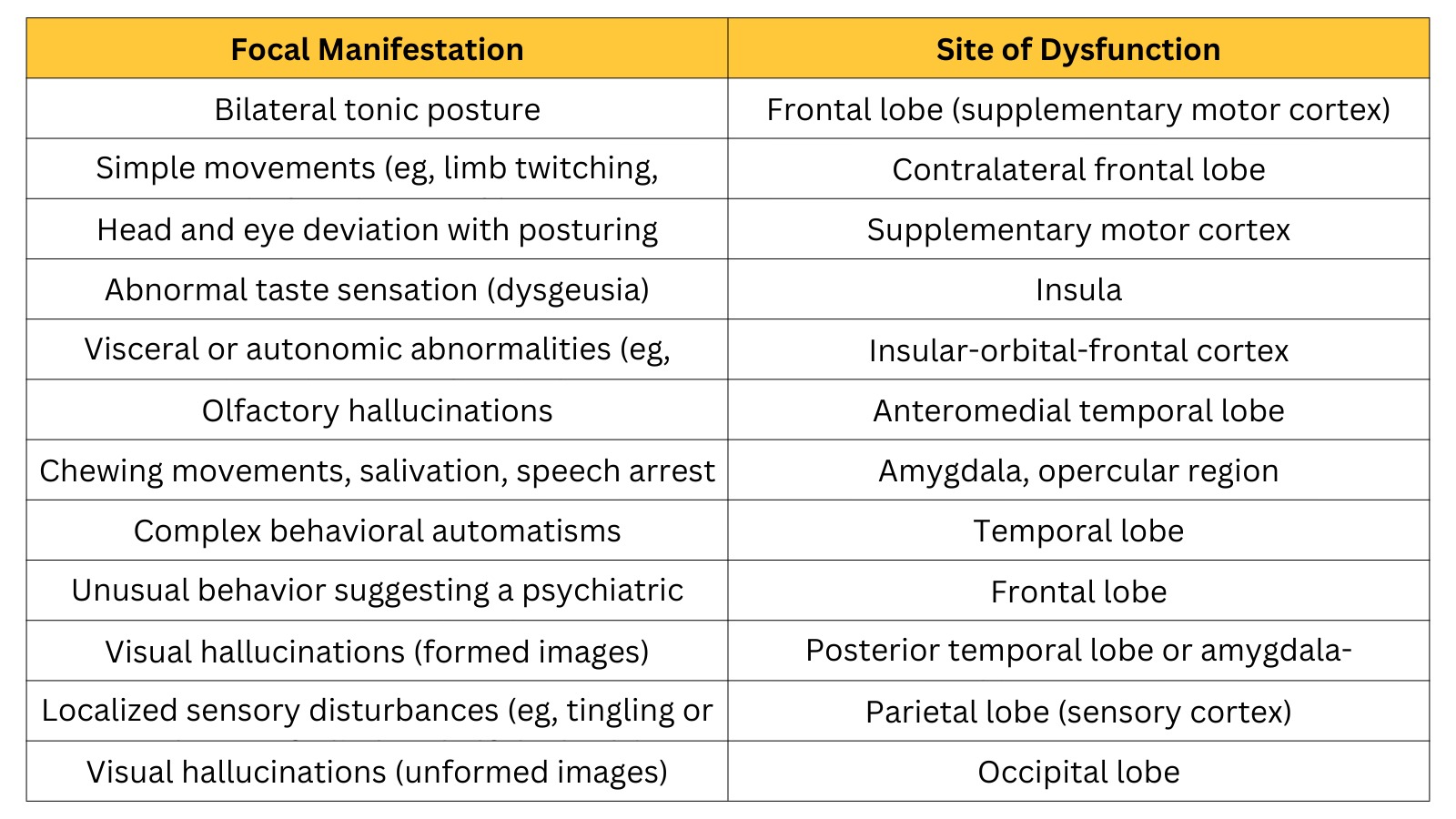
Table 1: Site of dysfunction according to manifestations of focal seizures
Generalized-Onset Seizures
Generalized-onset seizures start with a sudden loss of consciousness, so patients do not recall the seizures. Clinically, they could have motor or non-motor symptoms. Motor generalized seizures are tonic-clonic (grand-mal) seizures, myoclonic, atonic, tonic, or clonic seizures.
Tonic-clonic seizures have two phases. The tonic phase involves generalized muscle contractions; the eyes rotate, there could be apnea and cyanosis, lateral tongue biting, pooling of oral secretions, a loud moan–ictal cry, from contraction of the laryngeal and respiratory muscles, and urinary or fecal incontinence. This is accompanied by an increased sympathetic tone, so the pupils dilate, and the heart rate and blood pressure increase. The second phase is the clonic phase with rhythmic muscle twitching.
Clonic seizures involve rhythmic bilateral muscle contractions, in tonic seizures there’s muscle stiffening which may be unilateral or bilateral, myoclonic seizures have sudden jerky muscle twitching that is nonrhythmic and irregular, and atonic seizures (also called “drop seizures” or “drop attacks”) manifest with an abrupt loss of muscle tone leading to sudden head drop or collapse, they’re linked to epilepsy syndromes and are mistaken for syncope.
Generalized non-motor symptoms, also known as absence seizures, are a common feature of childhood epilepsy syndromes, usually beginning between ages 5-15 and do not continue into adulthood. Typical absence seizures, formerly known as petit mal seizures, involve a sudden onset of loss of consciousness, unresponsiveness, blank stare, and cessation of any motor activity. Patients do not convulse, axial muscle tone may or may not be lost, and subtle automatisms, eyelid flutter, or mild myoclonus, which often go unnoticed, like lip-smacking, eye fluttering, or head nodding, are common. The typical absence seizures last from 10 to 30 seconds, with an abrupt onset and offset of symptoms, as patients return rapidly to what they were previously doing without a post-ictal state or any memory of the seizure. Unfortunately, these seizures are sometimes mistaken for simple ‘daydreaming’ and the only complaint could be a poor school performance, made by the teacher. They’re classically triggered by hyperventilation and flashing lights, and if untreated, may occur many times a day.
Atypical absence seizures differ from typical ones in the following ways: they occur and subside more gradually, last longer, with more prominent, noticeable jerking and automatisms, and less complete loss of awareness and responsiveness. Unlike typical absence seizures, patients may have a history of damage to the nervous system, an abnormal neurological examination, and developmental delay, and these seizures may persist into adulthood.
Epilepsy syndromes
Infantile spasms/West syndrome is a rare, severe form of epilepsy typically beginning in the first year of life (peak onset: 3–7 months). Structural and metabolic causes are responsible for 70–80% of cases, such as hypoxic-ischemic encephalopathy, CNS malformations, tuberous sclerosis, infections, and metabolic disorders. It classically presents as sudden symmetric, synchronous contractions of the neck, torso, and extremities, usually in clusters of 5–10. These could be jerking flexions (jackknife movement) or extensions followed by a tonic phase. Hypsarrhythmia is a characteristic finding in interictal EEG, with high-voltage delta waves with irregular multifocal spikes, and slow waves. It carries a poor prognosis with increased mortality, neurodevelopmental delay, regression of psychomotor abilities, and most cases develop other forms of epilepsy, especially Lennox-Gastaut syndrome in 25% of cases. First-line treatment is ACTH, prednisone, or vigabatrin –particularly in tuberous sclerosis (18).
Lennox-Gastaut syndrome is a lifelong, severe developmental epileptic encephalopathy characterized by intellectual disability, multiple seizure types: myoclonic, tonic, atonic, absences, developmental delays, and frequent periods of status epilepticus. Most cases are due to structural brain abnormalities (tuberous sclerosis, meningitis, hypoxic-ischemic injuries, head injuries), while 40% are cryptogenic. The prognosis is unfortunately poor; only 10% are seizure-free under treatment, and many become resistant to medication, requiring other methods like ketogenic diet, vagus nerve stimulation, and surgery (19).
Juvenile myoclonic epilepsy (JME)/Janz syndrome is an idiopathic generalized epilepsy syndrome. It represents a triad of seizures that could happen in isolation or together: bilateral symmetrical myoclonic jerks, mainly following awakening and without impaired consciousness, generalized tonic-clonic seizures, and the least common of the three, absence seizures with impaired consciousness. Common seizure triggers include sleep deprivation, alcohol consumption, and flickering lights, and the peak incidence is around 12-20 years of age. Although it responds well to medications and seizures become less frequent in adulthood, there’s a high risk of recurrence, so lifelong treatment is usually required. An increased risk of psychiatric comorbidities has been reported with JME.
Dravet syndrome (also called Severe Myoclonic Epilepsy of Infancy, SMEI) is a developmental and epileptic encephalopathy that typically begins in the first year of life in otherwise healthy infants, characterized by recurrent seizures and poor neurodevelopment. Seizures are classically triggered by fever, infections, hot baths, and photic stimulation, and include prolonged febrile seizures (often the first seizure, lasting more than 15 minutes), myoclonic seizures, and sometimes focal and tonic-clonic seizures. Atypical absence and status epilepticus can appear later. It is most commonly caused by de novo mutations in the voltage-gated sodium channel alpha-1 gene (SCN1A) (20).
Benign epilepsy with centrotemporal spikes (BECTS) or Rolandic epilepsy is the most common form of epilepsy in children. BECTS is theorized to have a genetic cause, and although no gene has been identified, an autosomal dominant inheritance has been postulated. BECTS has a low frequency of seizures, low tendency to generalize, good prognosis, as most children become seizure-free by the age of 14, with normalization of the EEG and without neurological deficits –thus, termed benign, and the peak incidence is around 7–9 years of age. Ictal manifestations happen more frequently during NREM (Non-Rapid Eye Movement) sleep, mainly at sleep onset or just before awakening. The seizures are usually brief, lasting for 1-3 minutes, mainly affecting the face, oropharynx, with somatosensory and motor focal symptoms like speech arrest, hypersalivation, and, in some cases, upper limb involvement, and secondary generalization is observed. Characteristic EEG shows high-voltage spikes or spikes and waves in the centrotemporal region that may shift from side to side with a normal background, while neuroimaging is normal (21).
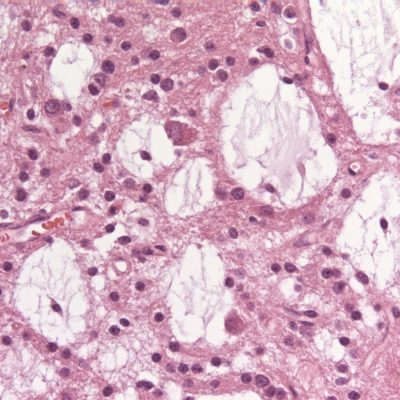
Temporal lobe epilepsy is the most common form of focal epilepsy. 70% of temporal lobe epilepsies arise from mesial temporal sclerosis/hippocampal sclerosis (Figure 4), which involves neuronal cell loss, gliosis, and sclerosis primarily in the dentate gyrus and CA1 section of the hippocampus, and loss of hippocampal volume and an increased T2 signal on MRI. Other causes of temporal lobe epilepsy include encephalitis (e.g., herpes simplex encephalitis), developmental disorders, neurodegenerative disorders, and tumors. Seizures occur in clusters lasting 30 seconds to 2 minutes on average. Typical symptoms include a visceral, olfactory, or auditory aura or feelings of familiarity or unfamiliarity, ictal motor symptoms –classically oral alimentary automatisms, or autonomic symptoms, and altered mental state without total loss of consciousness. With recurrent seizures, the limbic system can become impaired, resulting in impairment of libido, the menstrual cycle, and fertility, and memory. EEG shows temporal lobe spikes, and treatment includes pharmacotherapy, but due to the overall unfavorable prognosis of this syndrome, surgical options are offered for medication-resistant cases (22).
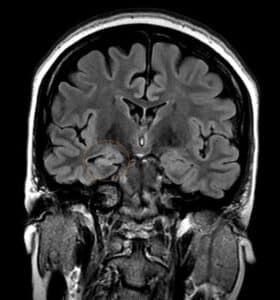
Figure 4: Right mesial temporal sclerosis
Febrile seizures
As mentioned above, febrile seizures are classified based on clinical features into simple febrile seizures, complex febrile seizures, and febrile status epilepticus. Simple febrile seizures are generalized-onset seizures –commonly tonic-clonic– that last less than 15 minutes, do not recur within 24 hours, and in the post-ictal phase, the patient does not experience fatigue with quick return to baseline. Febrile seizures are considered complex if they exhibit any of the following features: last more than 15 minutes, have a focal onset or focal features, recur in 24 hours, and have post-ictal neurological deficits or paralysis. So, a generalized febrile seizure lasting more than 15 minutes is classified as complex, and some sources even consider a febrile seizure in children with a pre-existing neurological deficit as complex (8).
A febrile status epilepticus may be focal or generalized. It is any febrile seizure that lasts for more than 30 minutes, or it could be a series of febrile seizures that last in total more than 30 minutes without a full recovery in inter-ictal phases.
Diagnosis
History and physical examination
Taking a comprehensive history from a seizure patient is an important skill, regardless of whether this is a first-time seizure or not. It is important to establish what has happened before, during, and after the event.
Before the event, establish any typical auras and suggestive features of the underlying etiology, like trauma or triggers that lower the seizure threshold. Other conditions –as will be discussed later– can mimic seizures, so a person must know the typical manifestations of the seizure mimics, such as palpitations before a cardiogenic syncope. A description of seizure semiology can be provided by either the patient if he was awake, any witnesses if the patient had an impaired consciousness, or other healthcare providers such as paramedics, and sometimes there would be video recordings. Identify the approximate duration of the seizure, and following the event, know how the seizure was terminated –through medications or spontaneously. Information about any prior seizures must be included, and risk factors for seizures and epilepsy should be assessed (23).
On examination, a patient may not have regained his or her consciousness fully yet; they may be confused, or have a bitten tongue, bruises from the event, or evidence of incontinence (e.g., urine or feces in clothing).
Workup
Testing is routinely done and depends on the specifics of the history and examination. However, the diagnosis of seizures and epilepsy remains majorly clinical, and so, normal or unchanged testing results do not exclude a seizure or epilepsy diagnosis (23,24).
Neuroimaging is recommended for first-time seizures and those with a prior history of seizures but had an abnormal examination for the first time but possibly avoided in a child with features suggestive of a characteristic seizure syndrome. CT detects any blood and masses, but provides limited visualization of brain anatomy and any pathologies. Thus, MRI remains superior for detecting any structural brain lesions like cortical or vascular malformations, traumatic gliosis, mesial temporal sclerosis, cerebral venous thrombosis, and tumors (23,24).
EEG may be the most definitive indication of a seizure and is performed in individuals with a first-time seizure, insufficient information for classifying the seizure, or seizures refractory to treatment. Characteristic findings on EEG can assist in making a diagnosis of epilepsy. However, the absence of EEG findings does not reliably rule out epilepsy, since an initial EEG identifies epileptiform abnormalities in only 30-55% of patients with a confirmed seizure disorder. Repeated EEGs, particularly those with prolonged recording durations or performed after sleep deprivation, significantly improve the likelihood of detecting such abnormalities in individuals with epilepsy. Some conditions are characterized by seizures with characteristic discharge patterns like hypsarrhythmia in West syndrome, and 3 Hz spike-and-wave in typical absence seizures (23,24).
Inpatient combined video-EEG monitoring can be performed for hospitalized patients, usually for 2 days. It incorporates the EEG recording with clinical behavior and is highly sensitive. Ambulatory EEG monitoring can be done at home and is useful if patients aren’t able to be admitted to the hospital for long periods of time (23,24).
In children with a typical febrile seizure with a normal neurological exam and a smooth return to baseline, neuroimaging and EEG can be deferred (23,24).
Laboratory screening is done for first-time seizures, or atypical manifestations, and a suspected metabolic, infectious, or toxic cause. They include serum electrolytes, blood urea nitrogen (BUN), creatinine, glucose, calcium, magnesium, and phosphate levels, and liver function tests. In patients with an established diagnosis, blood levels of antiseizure medications could be considered as well (23,24).
Other tests are guided by clinical suspicion, such as lumbar puncture for meningitis, drug screens, and ECG for cardiogenic syncope, though it is recommended for all patients with a reported loss of consciousness during a seizure (23,24).
Seizure imitators
The rates of epilepsy misdiagnosis are high worldwide, making it important to consider other paroxysmal events that could mimic a seizure. History remains key for a proper diagnosis, and if available, a video recording (25).
Vasovagal syncope is experienced by many across a wide age range. It’s often triggered by prolonged standing, dehydration, a change in posture, and emotional upset. The patient may experience dizziness and appear pale and have minimal post-event symptoms. Although the patient is typically flaccid, stiffening and tonic-clonic movements can be experienced by up to 50% of people, but are usually brief, lasting only seconds (25).
Cardiac syncope may be life-threatening, so it is important to recognize and manage it appropriately. It can be preceded by palpitations, and a standard ECG may be normal (25).
Strokes and transient ischemic attacks (TIA) manifest depending on the affected brain lesions, and the symptoms are commonly negative, like visual loss or weakness. TIAs last minutes to hours, while a stroke may last longer or have permanent deficits. The patient remains conscious throughout, and usually has predisposing factors like hypertension or hypercholesterolemia (25).
Breath-holding spells happen in pre-school children following an upset, long yell, or cry. This yell then stops in a prolonged expiratory apnea as the child’s face becomes cyanosed. Generalized stiffening and jerks are possible as it may trigger brain hypoxia and a subsequent seizure; still, recovery is rapid. They have a good prognosis but are shown to be more common in children with iron deficiency anemia (25).
Self-gratification includes normal behaviors that may be observed as early as infancy and up to school age. The event that is confused with a seizure commonly involves rhythmic hip flexion or adduction, accompanied by flushing and sweating, and a distant stare. The distant stare may confuse it with a focal impaired awareness seizure (25).
Panic attacks involve somatic symptoms like palpitations, chest pain, sweating, breathlessness, dizziness, fear of losing control, and/or dying, and have a variable duration. There’s sometimes a genetic predisposition and a history of an anxiety disorder (25).
Psychogenic non-epileptic spells (PNES) resemble epileptic seizures, especially focal aware or impaired awareness seizures, but have no evidence on EEG or clinical assessment of seizures. The event includes asynchronous muscle movement, sometimes with auras or vocalizations, and the patient is aware and can recall it. Tongue-biting, other forms of self-injury, or incontinence are uncommon. They’re more common in people with a history of psychological trauma, physical or sexual abuse, or a psychological disorder (25).
Benign neonatal sleep myoclonus is a harmless sleep phenomenon that starts in the neonatal period and persists for months. There are synchronous jerky movements of all limbs or less, and facial involvement is especially rare. These events typically last seconds, are separated by a variable length of time, and waking the child would abolish the movements. The neurological examination, feeding, and behavior are normal (25).
Migraine with visual aura is commonly confused with seizures; the patient experiences flashes, fortification spectra –arches of lights, specks, flames, or scotomas. Overall, migraines more commonly affect females, with a genetic predisposition, and could be triggered by stress or hormonal changes (25).
Spinal myoclonus manifests as a myoclonic jerk that may not be modified by sleep or by voluntary action, so it can be present when awake and during movement. Segmental spinal myoclonus hints at an underlying structural spinal pathology like syringomyelia and is limited to a few contiguous myotomes. Propriospinal spinal myoclonus involves axial muscles, is triggered by cutaneous stimuli, tendon reflexes, or recumbent position, and usually has no clear etiology (25).
Management
General principles in managing seizures include removal of the cause or provoking factors, if possible, which is the optimal approach; assessing the risk of recurrence in first-time seizures; and implementing precautions when the loss of consciousness during a seizure may be harmful.
If the risk of recurrence following a first-time seizure is high or a diagnosis of epilepsy is made, pharmacotherapy is often required. Non-pharmacological methods like surgery, nerve stimulation, and ketogenic diet may be offered for medication-refractory seizures, as will be further discussed later.
Pharmacotherapy and Antiseizure medications (ASMs)
Antiseizure medications (ASM) work by raising the seizure threshold, which is pathologically reduced in epilepsy patients (26).
Valproate works by inhibiting GABA transaminase, therefore increasing GABA levels, as well as inactivation both calcium and sodium channels. It has many adverse effects, including alopecia, weight gain, and hepatotoxicity (26).
Carbamazepine, phenytoin, fosphenytoin, and lamotrigine work by blocking sodium channels. Phenytoin is rarely used nowadays due to its numerous side effects, like hirsutism, gingival hyperplasia, osteopenia, and hepatotoxicity. Carbamazepine has side effects like hyponatremia from syndrome of inappropriate antidiuretic hormone secretion (SIADH) (26).
Topiramate blocks sodium channels, too, and potentiates the GABA receptors. Side effects include angle-closure glaucoma, weight loss, kidney stones, and cognitive problems (26).
Ethosuximide inhibits the T-type calcium channels in the thalamus and sometimes leads to gastrointestinal disturbances (26).
Phenobarbital –a barbiturate– and benzodiazepines are direct and indirect GABA-A receptor agonists, respectively. Sedation, dependence, and respiratory depression are expected with their use (26).
Tiagabine inhibits GABA reuptake; it’s an adjunctive to treatment of focal seizures and may cause dizziness (26).
Levetiracetam blocks SV2A receptors, modulating GABA and/or glutamate release, and inhibits voltage-gated calcium channels. Psychiatric adverse effects like personality changes have been reported with its use (26).
Gabapentinoids –pregabalin and gabapentin- are GABA analogs but do not bind to GABA receptors; rather, they exert their effect by inhibiting presynaptic P/Q-type calcium channels and reducing glutamate release (26).
Vigabatrin irreversibly inhibits GABA transaminase and is only used in patients if other anticonvulsants are ineffective, as irreversible vision loss occurs in up to 50% of patients (26).
Skin reactions like Stevens-Johnson syndrome (SJS) and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported with the use of ASM, especially lamotrigine, carbamazepine, phenobarbital, fosphenytoin, and phenytoin induce them (26).
Some ASMs can interfere with the effects of other drugs through the cytochrome P450 enzymes; valproate inhibits them, while phenobarbital, carbamazepine, phenytoin, and fosphenytoin cause induction (26).
ASMs have a high risk of teratogenicity; the risk is generally lower with monotherapy but differs by drug. Higher-risk drugs include phenobarbital, topiramate, carbamazepine, phenytoin, and valproate. Some newer drugs, like lamotrigine and levetiracetam, appear to carry lower risks (26).
The Choice of medication to treat seizures
No single medication treats all seizures, and the choice depends on the seizure and epilepsy type, ASM side effects, patient’s age, comorbidities, and childbearing plans in women. Monotherapy is always preferred, but if seizures are poorly controlled, another drug can be added. When combining ASM, physicians often use agents with different mechanisms of action; usually, 2-3 medications can be administered safely in these patients (27-29).
Broad-spectrum AEDs are effective for generalized and focal onset seizures and include lamotrigine, levetiracetam, valproate, zonisamide, and topiramate (27-29).
For generalized onset tonic-clonic seizures, valproate and divalproex are the first line of treatment, lamotrigine and levetiracetam are second, and zonisamide can be used as an adjunctive medication (27-29).
Typical absence seizures are treated with ethosuximide or valproate as the first line of treatment, while lamotrigine comes second, and many children are seizure-free by adulthood. Atypical absence seizures, on the other hand, are treated with valproate, lamotrigine, or topiramate (27-29).
Myoclonic and atonic seizures are treated with valproate or vigabatrin. Infantile spasms are treated with ACTH, corticosteroids, or vigabatrin (27-29).
First-line options for the different types of focal-onset and focal-to-bilateral seizures include carbamazepine, fosphenytoin, lamotrigine, levetiracetam, oxcarbazepine, phenytoin, and topiramate (27-29).
Juvenile myoclonic epilepsy responds well to valproate or divalproex but requires lifelong therapy due to the high risk of recurrence (27-29).
Lennox-Gastaut syndrome unfortunately responds poorly to medications, including e.g., valproic acid, clobazam, lamotrigine, felbamate, and may often require surgery or neurostimulation (27-29).
Rolandic epilepsy /Benign epilepsy with centrotemporal spikes (BECTS) are offered ASM in case of high seizure frequency or severity, such as valproate, and many patients will have their seizures resolved by puberty (27-29).
For febrile seizures, administering ASM is not recommended and may lead to many medications related problems. If the patient develops another seizure in the absence of fever, however, ASM could be considered (27-29).
Nonpharmacological therapy
Nonpharmacological therapy is mainly indicated for pharmaco-resistant epilepsy and includes surgical interventions, stimulation techniques, and dietary measures (30).
Surgical options involve resection or disconnection procedures. Resection entails the removal of pathological lesions, such as anteromedial temporal lobe or hippocampal resection in patients with temporal lobe epilepsy due to hippocampal sclerosis, or hemispherectomy in cases of severe intractable seizures confined to one hemisphere. Disconnection surgeries include corpus callosotomy, where the corpus callosum is sectioned—initially the anterior two-thirds, with complete disconnection considered if seizures persist. Another disconnection procedure is hemispherotomy, which disconnects the cortex of one hemisphere from ipsilateral subcortical structures and the contralateral cortex without removing the hemisphere itself (30).
Stimulation techniques provide another option, such as vagus nerve stimulation, where intermittent electrical impulses are delivered to the left vagus nerve through a pacemaker-like device that patients can activate with a magnet to abort seizures. Deep brain stimulation involves electrodes placed near specific deep brain structures connected to a chest wall generator, while responsive neurostimulation uses an intracranially implanted device that detects epileptiform activity and directly stimulates the seizure focus to prevent progression into a seizure (30).
Additionally, dietary interventions like the ketogenic diet, although supported by limited evidence, have shown effectiveness in reducing seizure frequency, particularly in children with syndromes such as West or Dravet (30).
Acute management of seizures and status epilepticus
Initial stabilization of acute seizures begins with calling for help, ensuring patient safety by removing nearby hazards, and performing an ABCDE assessment with cardiopulmonary resuscitation if needed. Basic airway maneuvers should be initiated, oxygen therapy started, and the patient placed in the recovery position, followed by checking point-of-care glucose and vital signs.
Pharmacologic management depends on seizure duration: early seizures (0–5 minutes) are often self-limited and usually do not require medication; early status epilepticus (5–20 minutes) is treated with IV benzodiazepines (lorazepam or diazepam) or IM/intranasal/buccal/rectal alternatives, repeated if necessary; persistent status epilepticus (20–40 minutes) requires second-line agents such as IV fosphenytoin, valproic acid, or levetiracetam, with phenobarbital as an alternative; and refractory status epilepticus (40–60 minutes) may necessitate repeating second-line therapy or inducing coma with agents like propofol, thiopental, midazolam, or pentobarbital. Throughout, reversible causes such as hypoglycemia, hyponatremia, or hypocalcemia must be promptly corrected (31).
Prognosis and complications
After a first unprovoked seizure, the risk of recurrence is approximately 40–50% within two years in untreated individuals, with 80% of recurrences occurring within that two-year window. If there is no underlying brain insult—such as stroke, trauma, or CNS infection—this risk remains at the same 40–50% level. Following a brain insult occurring at least one week prior to the seizure (i.e., an acute symptomatic situation), recurrence risk over the next 10 years increases to around 65%. After a second unprovoked seizure, the risk of another seizure jumps to approximately 60% within one year. In contrast, the recurrence risk following an acute symptomatic (provoked) seizure is lower—about 19% over the next 10 years (32)
Treatment outcomes are encouraging; about 60–70% of treated patients remain seizure-free at 10 years after the first seizure. When it comes to withdrawal of treatment after sustaining a two-year seizure-free period on antiepileptic drugs, approximately 60–90% of children and 35–57% of adults remain seizure-free following discontinuation of therapy.
About 30–40% of people with epilepsy develop drug-resistant epilepsy (DRE) despite multiple treatment attempts. The probability of achieving seizure freedom with further medication trials is <5% per additional drug tried. DRE is associated with reduced quality of life, cognitive and psychiatric comorbidities, increased health-care utilization, and higher risk of injury and premature death, including SUDEP(33).
Regarding mortality, individuals with epilepsy face a 1.6–3-fold higher risk of all-cause mortality compared to the general population. Sudden Unexpected Death in Epilepsy (SUDEP), a dreaded complication, has an incidence ranging from about 1.2 to 6.3 per 1,000 individuals with epilepsy per year—higher in those with refractory or chronic epilepsy. SUDEP accounts for an estimated 8–17% of all epilepsy-related deaths (34).
Complications
Epilepsy is associated with both acute and long-term complications. Acute complications include hyperthermia, cardiorespiratory disturbances, and excitotoxicity, which may cause irreversible central nervous system injury such as cortical laminar necrosis, thereby increasing the risk of future seizures. Postictal metabolic changes are also common, particularly transient lactic acidosis characterized by elevated lactic acid and decreased serum bicarbonate, which typically resolves spontaneously within 60–90 minutes after seizure cessation. Additionally, seizures may result in physical trauma, such as tongue biting, posterior glenohumeral dislocation from falls, polytrauma in accidents, or prolonged convulsive episodes leading to status epilepticus (35).
The long-term consequences of epilepsy extend beyond seizure activity itself and include a range of psychiatric disorders—such as anxiety, depression with elevated suicide risk, cognitive decline, psychosis (whether interictal, postictal, or medication-induced), and psychosocial stressors, including occupational challenges. Sleep disturbances and insomnia are also prevalent, alongside bone disease (osteomalacia and osteoporosis), often secondary to chronic antiseizure medication use.
A particularly feared outcome is Sudden Unexpected Death in Epilepsy (SUDEP), defined as the sudden, unexplained death of a person with known epilepsy in the absence of trauma, drowning, or other medical conditions. SUDEP most often occurs during sleep and is more frequent in patients with intractable epilepsy, frequent generalized tonic–clonic seizures, and those with early disease onset (6,36).