Article Topic: Limb Girdle Muscular Dystrophy.
Author: Bassel Qiqieh.
Editors: Haneen Al-Abdallat, Haneen A. Banihani.
Reviewers: Rahmeh Adel, Ethar Hazaimeh
Keywords: Limb girdle dystrophy, caplain-3, sarcoglycan, dystroglycanopathy, Dysferlinopathy.
Abstract
Limb-girdle muscular dystrophies (LGMDs) constitute a group of genetic muscle disorders characterized by progressive weakness and atrophy of proximal muscles. They result from mutations in genes encoding subtype-specific proteins
with autosomal inheritance patterns. In 2018, a refined definition of LGMDs was proposed by the European Neuromuscular Centre. Classification of LGMDs has evolved to include the mode of inheritance, genetic discovery order, and the affected protein.
Epidemiologically, prevalence varies globally, with the recessive form being more common. Notably, Calpainopathies are the most prevalent subtype. Pathophysiology of LGMDs involves diverse molecular pathways, including the dystrophin-glycoprotein complex, the sarcomere, and signal transduction pathways.
Clinical manifestations of LGMDs range from childhood to adulthood onset, with common symptoms including muscle weakness, muscle cramps, fatigue, and exercise intolerance. Common complications include Rhabdomyolysis and cardiopulmonary involvement. Diagnosis is challenging due to the overlap in symptoms. Due to recent advancements in Next-Generation Sequencing (NGS), effective diagnosis of LGMD is faster and more cost-effective.
Management primarily involves rehabilitation and multidisciplinary care to prevent complications. Research into molecular and gene therapies, such as stem-cell transplantation and exon skipping, offers promising avenues for treatment. However, challenges persist, particularly in interpreting genetic variants of uncertain significance (VUSs).
In conclusion, LGMDs represent a diverse group of disorders with significant clinical and genetic heterogeneity. Advances in diagnostic techniques and therapeutic methods will help further improve patient outcomes and understanding of these conditions.
Overview
Limb-girdle muscular dystrophies (LGMDs) rank as the fourth most prevalent type of genetic muscle disorders [1], constituting a diverse set of inherited conditions marked by progressive weakness and atrophy of proximal muscles [2].
These conditions arise due to mutations in genes that encode subtype-specific proteins. The inheritance pattern of LGMD is autosomal, differing from Duchenne or Becker muscular dystrophies. LGMD can exhibit both dominant and recessive forms [3].
In 2018, the definition of LGMDs underwent further refinement by the European Neuromuscular Centre, and a new nomenclature was suggested [4].
The new current proposed definition of LGMD is: “A genetically inherited condition that primarily affects skeletal muscle leading to progressive, predominantly proximal muscle weakness at presentation caused by a loss of muscle fibers. To be considered a form of limb-girdle muscular dystrophy, the condition must be described in at least two unrelated families with affected individuals achieving independent walking, must have an elevated serum creatine kinase activity, must demonstrate degenerative changes on muscle imaging over the course of the disease, and have dystrophic changes on muscle histology, ultimately leading to end-stage pathology for the most affected muscles. ”[5]
The following article discusses the Classification, Epidemiology, Clinical manifestations, Pathophysiology, Diagnosis, Management and Treatment of LGMD.
Classification
In the previous classification system, the numbers ‘one’ or ‘two’ following LGMD described the inheritance type, with ‘one’ indicating dominant inheritance and ‘two’ indicating recessive inheritance. Additionally, a letter was assigned based on the order of genetic linkage discovery. Over 30 LGMD genetic subtypes have been identified, with the latest autosomal recessive addition labeled as LGMD2Z [5]. However, certain limitations have arisen from this previous classification: Firstly, the alphabet for LGMD2 has been fully utilized, including the recent addition of LGMD2Z. Secondly, there are other genetically characterized muscle diseases inherited autosomally that clinically resemble LGMD but are not classified as such. Thirdly, specific mutations in genes associated with LGMD can lead to both LGMD1 and LGMD2. Fourthly, certain LGMDs are also categorized as hereditary myopathies of other types [2].
As a result, the proposed formula for classifying subtypes of LGMD according to Straub [5] is “LGMD, inheritance (R or D), order of discovery (number), affected protein.” An example of this would be: “LGMD R1 Calpain3- related”.
Table 1 below shows a comparison between the old classification and the proposed one according to the new definition [5].
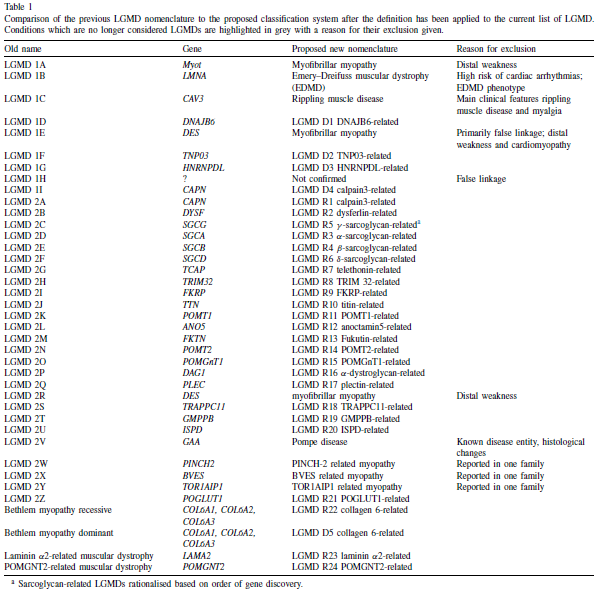
Table 1: Comparison of the previous LGMD nomenclature to the proposed classification system.5
Epidemiology
The prevalence of LGMD varies globally, with estimates ranging from 2.27-6.89 per 100,000. In the northern United Kingdom, LGMD makes up 6.15% of the neuromuscular population. The recessive form of LGMD is more common than the dominant one [6].
Calpainopathies (specifically LGMD R1 Calpain 3–Related) are the most prevalent among LGMD subtypes, accounting for 30–40% of LGMD cases, with an estimated worldwide prevalence of 6.8–10.2 cases per million. Typically, around 80% of patients experience wheelchair dependency between the second and fourth decades of life. Sarcoglycanopathies (specifically R3-R6) represent the most severe type of LGMD, comprising 10–25% of LGMD cases. The prevalence of sarcoglycanopathies varies across different ethnicities and geographic regions, ranging from 0.31 to 0.58 cases per 100,000, with most children presenting with progressive muscle weakness during childhood, which will later result in ambulation in adolescence [7]. LGMD R2 Dysferlin-Related is the most common subtype in Asia [4].
Pathophysiology
The underlying pathophysiology and molecular mechanisms in each subtype of LGMD are different. The pathways implicated in these conditions include the dystrophin-glycoprotein complex, the sarcomere, glycosylation of dystroglycan, vesicle, and molecular trafficking, signal transduction pathways, and nuclear functions [8].
Calpainopathies result from a deficiency in CAPN3. In typical physiological processes, calcium activation triggers Calpains, a family of Ca-dependent lysosomal cysteine proteases that play a significant role in cell motility, apoptosis, and cell differentiation. A lack of CAPN3 is associated with multiple effects on skeletal muscle, including oxidative damage, dysregulation of Ca2+, disorganization of sarcomeres, abnormalities in mitochondrial function, altered muscle adaptation, and impaired muscle regeneration. Collectively, these factors contribute to inflammation, necrosis, fibrosis, atrophy, and the progressive degeneration of muscle tissue [9].
A-Dystroglycanopathies arise from the abnormal glycosylation of a-dystroglycan. This heavily glycosylated sarcolemmal glycoprotein plays a pivotal role in maintaining sarcolemmal integrity by connecting the dystrophin-associated glycoprotein complex to the extracellular matrix. Consequently, this condition manifests as a wide range of symptoms, including muscle pseudohypertrophy, cardiac involvement, and cognitive impairment [2].
Sarcoglycanopathies comprise four subtypes of Limb-Girdle Muscular Dystrophy (LGMDR3-R6), that are derived from recessive mutations in the SGCA, SGCB, SGCG, and SGCD genes. These genes encode α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan, and δ-sarcoglycan, respectively. These proteins are integral components of the dystrophin-glycoprotein complex (DGC) found in the muscle sarcolemma. Mutations in these genes lead to a deficiency in their respective protein subunits, disrupting the closely linked heterotetramer complex associated with the dystrophin-associated protein complex in the cell membrane [7].
Dysferlinopathy and anoctaminopathy-5 stand out as prototypes among muscular dystrophies characterized by a primary impairment in membrane repair. These conditions arise from mutations in the genes encoding dysferlin (DYSF) and anoctamin-5 (ANO5), respectively. When the sarcolemma is disrupted, it prompts the recruitment of vesicles to the injury site, facilitated by a muscle-specific protein called Mitsugumin-53 (MG53). Dysferlin plays a crucial role in the fusion of these vesicles, forming a membrane repair patch. While the significance of anoctamin-5 in membrane repair has been established, the precise mechanism by which it operates within the membrane repair complex remains unclear [2].
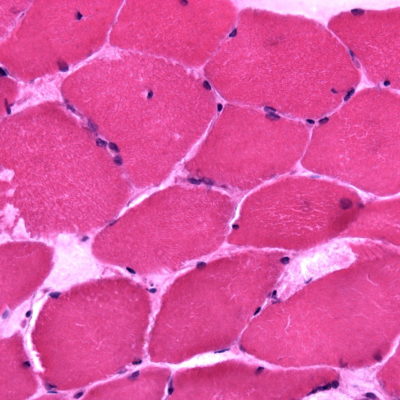
Histology
Figure 1 shows the histology of various subtypes of LGMDs [2].
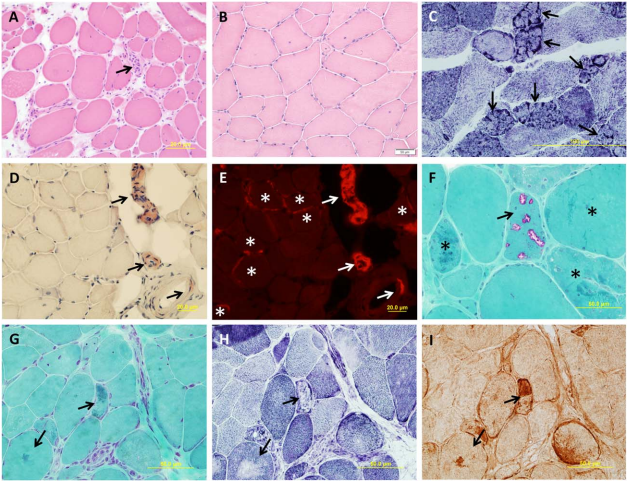
FIGURE 1. Muscle pathology of muscular dystrophies (A–I). (A) A laminopathy patient shows dystrophic features consisting of
necrotic fibers (arrow), varying fiber size, an increase in internal nuclei, fiber splitting, and interstitial fibrosis (hematoxylin and eosin).(B) A patient with dominant calpainopathy shows minimal nonspecific myopathic changes, consisting of variations in fiber size and an increase of internal nuclei (hematoxylin and eosin stain). (C) A patient with recessive calpainopathy demonstrates numerous lobulated fibers (arrows) (nicotinamide adenine dinucleotide stain). (D–E) Congo red–stained section reveals amyloid deposits in intramuscular blood vessel walls (arrows) and in scattered endomysial sites (asterisks) in a patient with dysferlinopathy. Amyloid deposits, especially those in endomysium, are better visualized under rhodamine optics (E) than light microscopy (D). (F) A patient with myofibrillar myopathy shows rimmed vacuoles (arrow) and fibers harboring amorphous hyaline materials (asterisks) (modified Gomori trichrome). (G–I) Consecutive sections from another patient with myofibrillar myopathy show amorphous hyaline materials on modified Gomori trichrome–stained section (G) (arrows), which correspond to areas lacking oxidative enzyme activity shown on nicotinamide adenine dinucleotide staining (H) (arrows). These amorphous materials are immunoreactive for aB-crystallin (I) (arrows).
Clinical manifestations
LGMD can present in early childhood, adolescence, and even in adulthood. All muscular dystrophies share skeletal muscle weakness as a common characteristic, but the distribution, extent, and progression of this weakness vary among different disorders [10].
The symptoms of LGMD often begin with challenges in walking upstairs and experiencing frequent falls [6]. However, for many individuals, the condition progresses to a loss of ambulation, typically occurring 10 to 20 years after the onset of the disease. Nevertheless, patients with mild forms of LGMD, especially those affected early in childhood, may retain their ability to walk [11].
Common Symptoms in Muscular Dystrophies [2]
- Muscle pain.
- Muscle cramps.
- Weakness especially around hip and shoulder girdle muscles.
- Fatigue.
- Exercise intolerance.
- Symmetrical muscle loss.
- Waddling gait.
- Winged scapula.
- Hyperlordosis
- Muscle contractures, especially in the Achilles tendon.
- Calf hypertrophy [12].
The distinctive features of each type of LGMD are listed in Table 2: [2]
| Type of LGMD | Distinctive features |
| Calpainopathies |
|
| a-Dystroglycanopathies | Encompass a wide spectrum of disorders affecting muscles, eyes, and the brain. This range spans from:
|
| Dysferlinopathy and anoctaminopathy-5 |
|
| Sarcoglycanopathies |
|
Complications
Rhabdomyolysis stands as a prevalent complication across all LGMD subtypes. Certain subtypes of LGMD are frequently associated with cardiopulmonary involvement. Notably, cardiomyopathies emerge as a common complication in Dystroglycanopathies and Sarcoglycanopathies. Among these, Sarcoglycanopathies exhibit the highest severity and pose the greatest risk of cardiomyopathies, representing approximately 30% of cases. However, such complication is rare in Calpainopathies and dysferlinopathies [2].
Diagnosis
Diagnosing LGMDs is challenging due to overlapping symptoms and genetic diversity. While specific clinical signs can help guide diagnosis, they are not definitive [6].
Similar to other hereditary conditions, the diagnosis of LGMD starts with a thorough review of medical and family history, coupled with a physical examination. Supportive diagnostic measures include: laboratory tests, such as assessing creatine kinase levels, conducting muscle magnetic resonance imaging, and utilizing electrodiagnostics. An MRI usually shows a consistent fatty infiltration pattern, irrespective of LGMD phenotype [13].
Figure 2 shows the MRI of an LGMD patient [14].
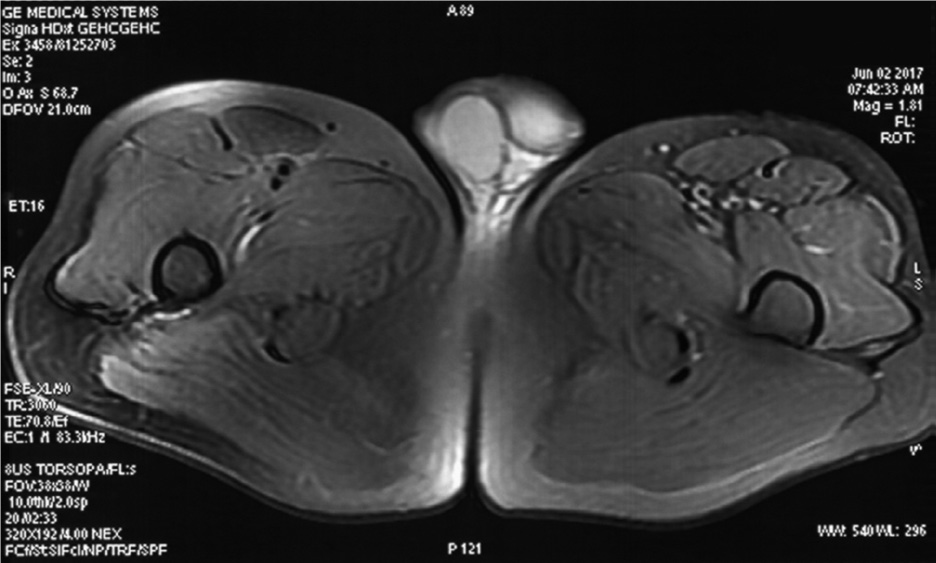
FIGURE 2. Magnetic resonance imaging (MRI) of the lower extremities. Images were acquired at the level of the upper femur. The T2WI fat suppression sequence showed slight atrophy and suspicious inflammatory changes of the left lateral femoral muscle and tensor fascia lata.
A conclusive diagnosis typically involves a muscle biopsy, processed through immunohistochemistry. It often reveals dystrophic changes, showcasing degeneration and regeneration of muscle fibers in the majority of LGMD cases [6]. Other findings include reduced levels of alpha-dystroglycan [12]. In calpainopathies, a muscle biopsy may show observable eosinophilic infiltrates, sometimes accompanied by elevated levels of eosinophils in the peripheral blood, resembling eosinophilic myositis in its early stages. However, these findings are not specific [2].
Nevertheless, due to the substantial phenotypic diversity and potential overlap among LGMD subtypes, precise determination of the LGMD genetic subtype necessitates genetic testing. The introduction of Next-Generation Sequencing (NGS) has significantly shortened the time required to sequence the entire human genome, decreasing from years to days. Moreover, the cost has substantially decreased from several billion dollars to a few thousand dollars [8].
Nowadays, panel-based NGS is being widely used in diagnosis, predicting, and assessing the risk of LGMD. Whole Exome Sequencing (WES), a strategy within Next-Generation Sequencing (NGS), has been documented as an effective approach when coupled with targeted analysis. This methodology has proven successful in uncovering disease-causing mutations that were previously overlooked through incomplete Sanger sequencing, increasing the success rate of diagnosing skeletal muscle disorders [1].
Management and Treatment
The primary care for individuals with LGMDs involves rehabilitation and preventing complications in other organ systems. Recommendations include seeking medical attention in neuromuscular clinics, consulting specialists in cardiology, pulmonary, and orthopedics, engaging in physical and occupational therapy, using orthotic and durable medical equipment, undergoing genetic testing with counseling, and encouraging an active and fulfilling lifestyle [4].
Current Research
The development of molecular and gene therapies for treating LGMD is currently in progress. Those strategies include Stem-cell transplantation, Exon skipping, gene delivery, RNAi, and gene editing [3]. Table 3 discusses Molecular and gene therapies studied for LGMD.
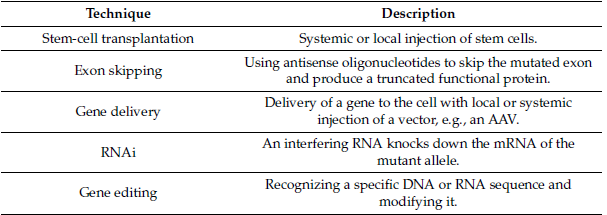
Table 3: Molecular and gene therapies studied for LGMD.
Moreover, advancements in Next-generation sequencing (NGS) has revealed numerous genetic variants within LGMD-related genes, many of which are categorized as variants of uncertain significance (VUSs). VUSs represent genetic alterations whose pathological implications remain unknown, posing significant challenges in both genetic test interpretation and disease diagnosis. Gaining insight into the phenotypic consequences of VUSs holds the potential to offer valuable clinical direction concerning LGMD risk assessment and therapeutic interventions [1].
Further reading
- Calcium mechanics in Limb-Girdle Muscular Dystrophies with CAPN3 mutations [9].
- Current and future approaches to classify VUS in LGMD-Related genes [1].
- Limb-Girdle Muscular Dystrophies Classification and Therapies [3].
- Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches [11].
References